

Prevalence and properties of intragenic copy-number variation in Mendelian disease genes
- PMID: 29895855
- PMCID: PMC6752305
- DOI: 10.1038/s41436-018-0033-5
Prevalence and properties of intragenic copy-number variation in Mendelian disease genes
Abstract
Purpose: We investigated the frequencies and characteristics of intragenic copy-number variants (CNVs) in a deep sampling of disease genes associated with monogenic disorders.
Methods: Subsets of 1507 genes were tested using next-generation sequencing to simultaneously detect sequence variants and CNVs in >143,000 individuals referred for genetic testing. We analyzed CNVs in gene panels for hereditary cancer syndromes and cardiovascular, neurological, or pediatric disorders.
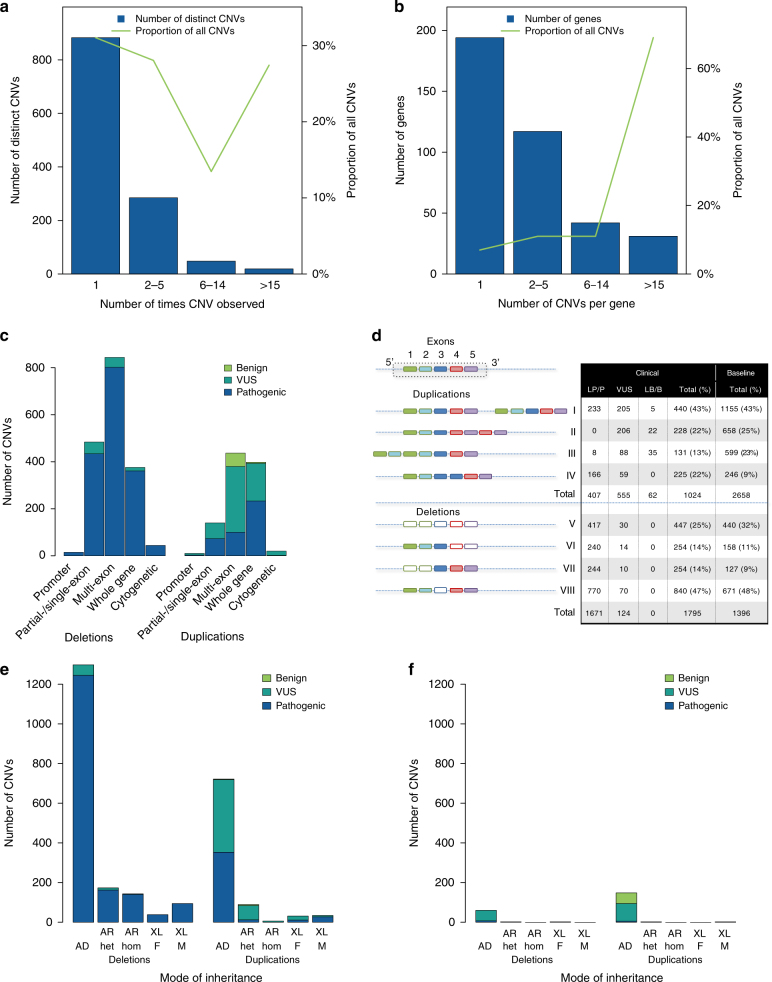
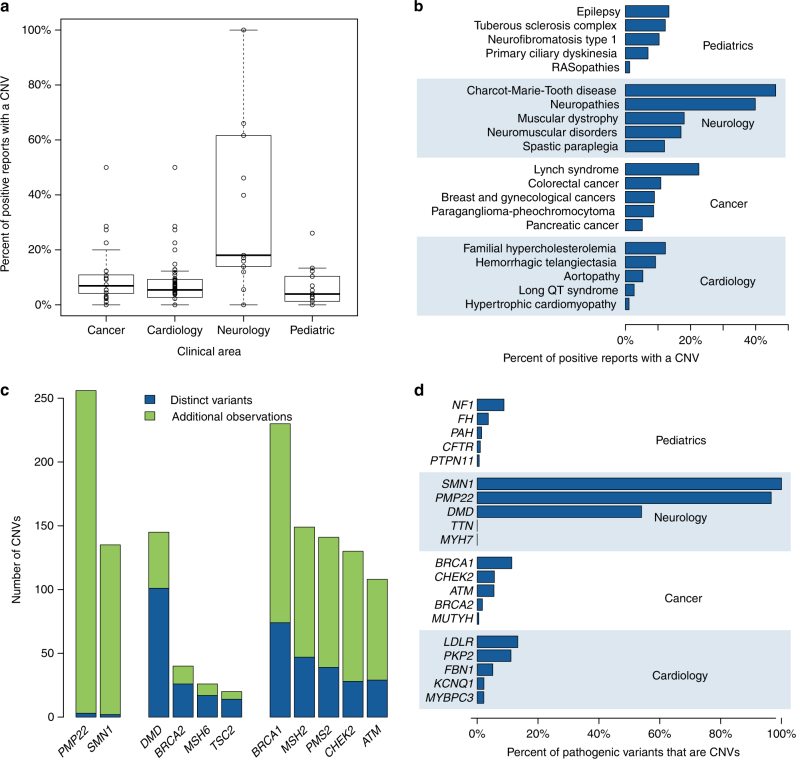
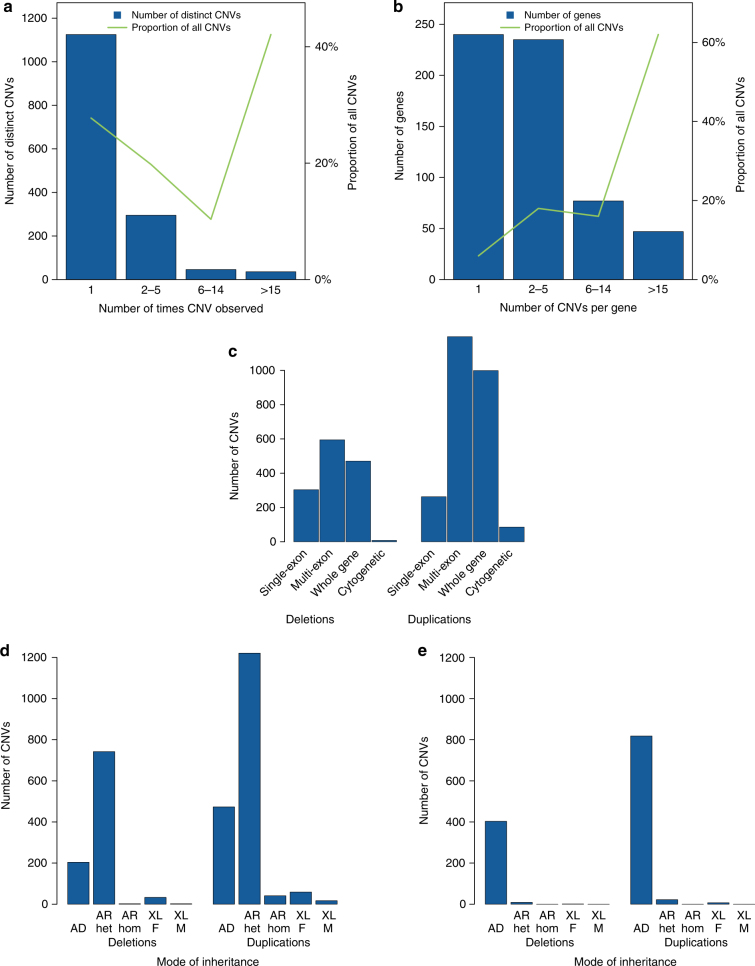
Results: Our analysis identified 2844 intragenic CNVs in 384 clinically tested genes. CNVs were observed in 1.9% of the entire cohort but in a disproportionately high fraction (9.8%) of individuals with a clinically significant result. CNVs accounted for 4.7-35% of pathogenic variants, depending on clinical specialty. Distinct patterns existed among CNVs in terms of copy number, location, exons affected, clinical classification, and genes affected. Separately, analysis of de-identified data for 599 genes unrelated to the clinical phenotype yielded 4054 CNVs. Most of these CNVs were novel rare events, present as duplications, and enriched in genes associated with recessive disorders or lacking loss-of-function mutational mechanisms.
Conclusion: Universal intragenic CNV analysis adds substantial clinical sensitivity to genetic testing. Clinically relevant CNVs have distinct properties that distinguish them from CNVs contributing to normal variation in human disease genes.
Keywords: Diagnostic genetic testing; Intragenic deletion/duplication copy-number variant; Next-generation sequencing panel; Pathogenic variation prevalence; Structural variant.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
