

Development Of A Three-Gene Prognostic Signature For Hepatitis B Virus Associated Hepatocellular Carcinoma Based On Integrated Transcriptomic Analysis
- PMID: 29896284
- PMCID: PMC5995946
- DOI: 10.7150/jca.23762
Development Of A Three-Gene Prognostic Signature For Hepatitis B Virus Associated Hepatocellular Carcinoma Based On Integrated Transcriptomic Analysis
Abstract
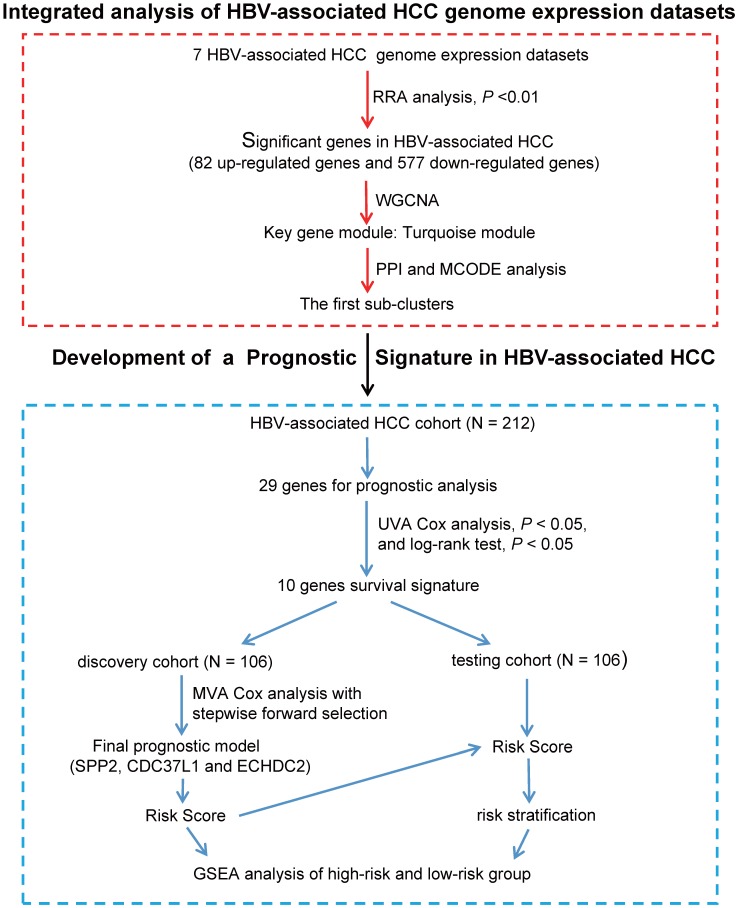
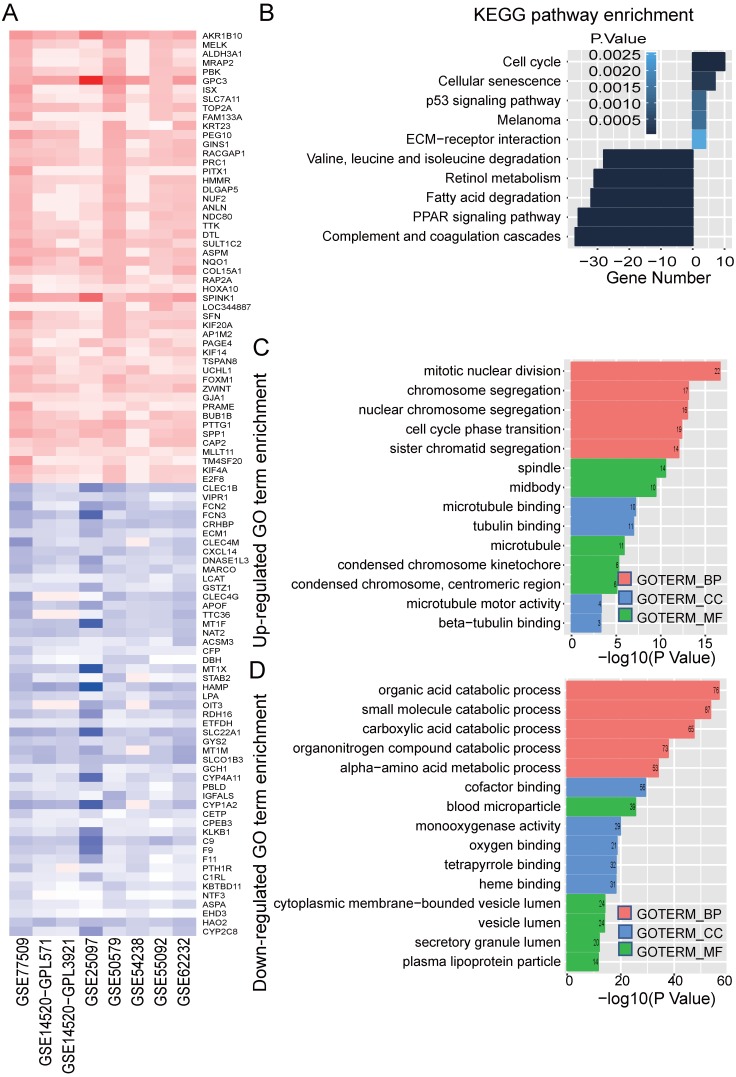
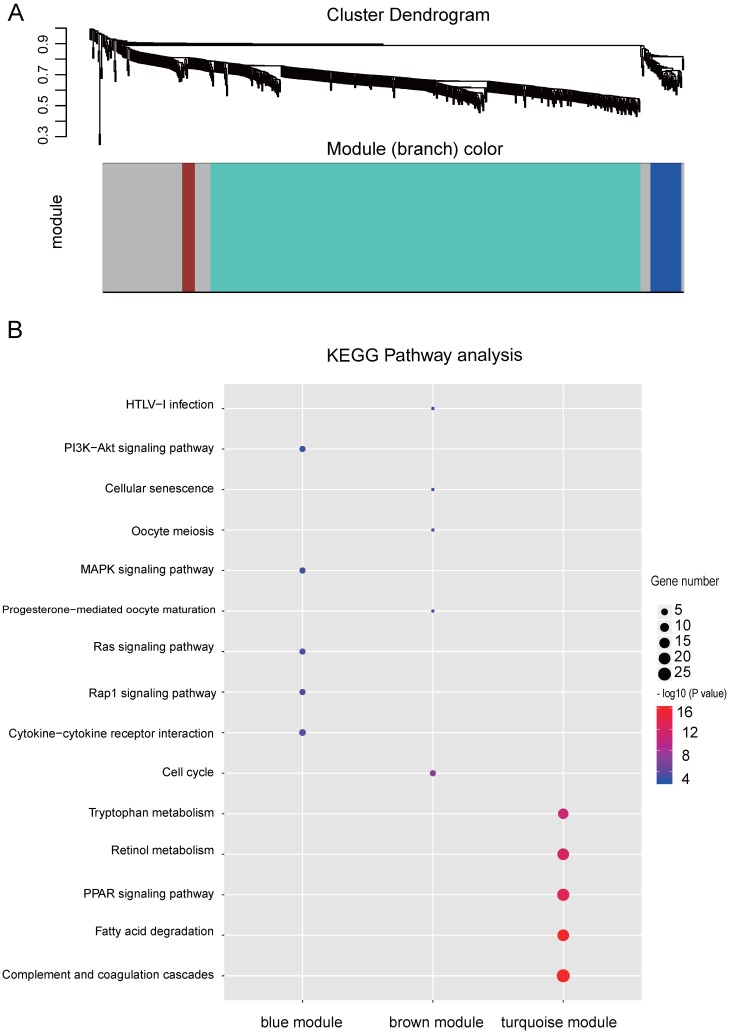
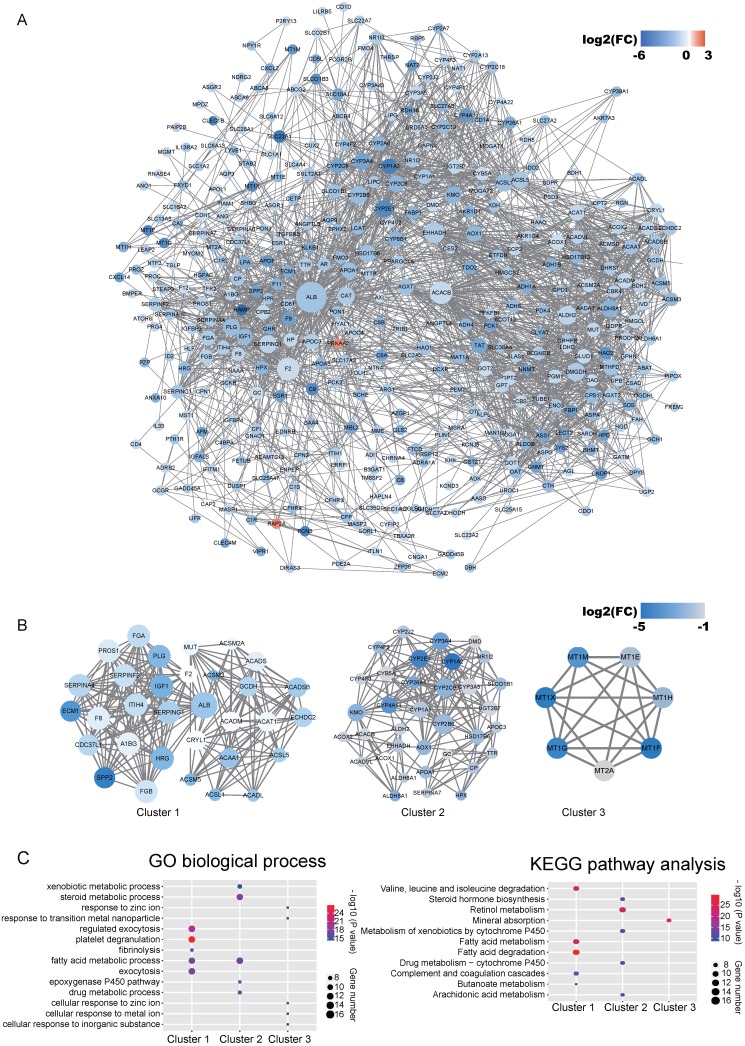
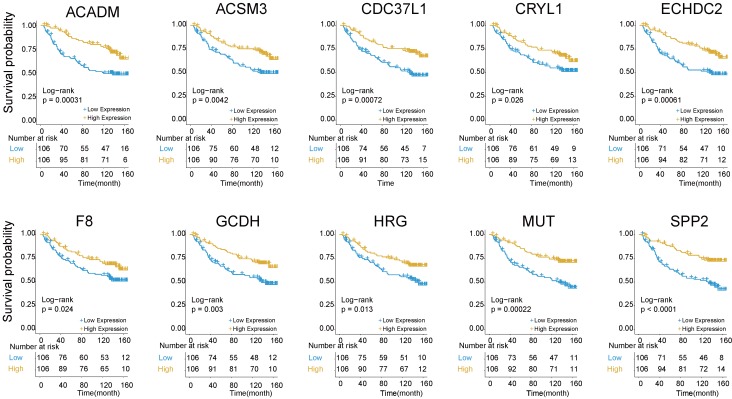
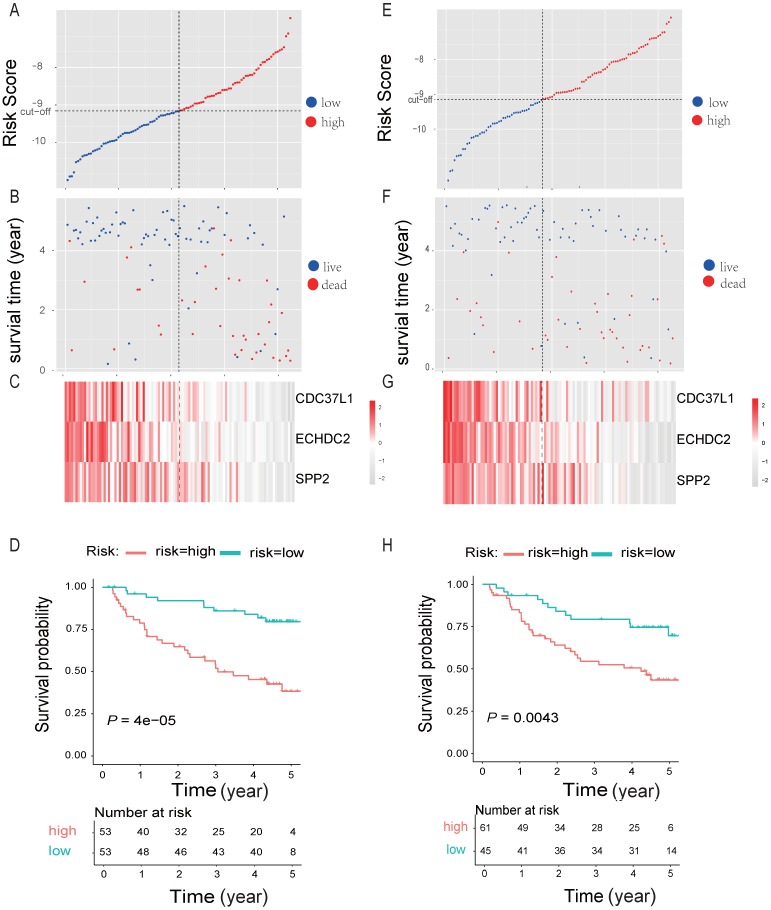
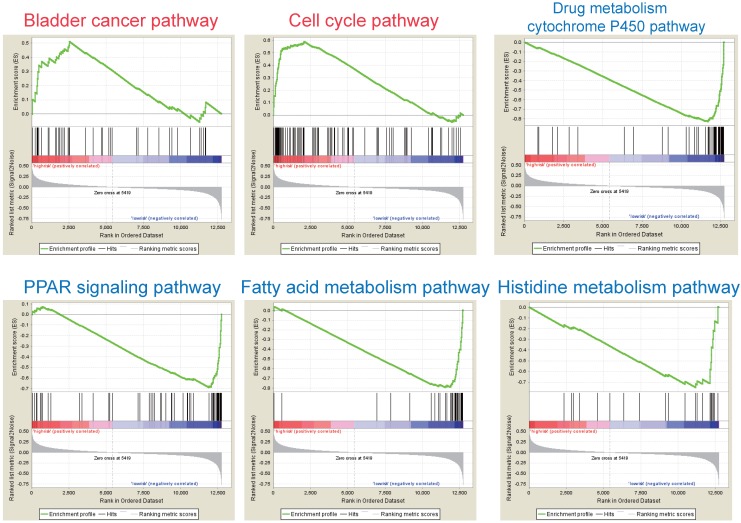
Integration of public genome-wide gene expression data together with Cox regression analysis is a powerful weapon to identify new prognostic gene signatures for cancer diagnosis and prognosis. Hepatitis B virus (HBV) is a major cause of hepatocellular carcinoma (HCC), however, it remains largely unknown about the specific gene prognostic signature of HBV-associated HCC. Using Robust Rank Aggreg (RRA) method to integrate seven whole genome expression datasets, we identified 82 up-regulated genes and 577 down-regulated genes in HBV-associated HCC patients. Combination of several enrichment analysis, univariate and multivariate Cox proportional hazards regression analysis, we revealed that a three-gene (SPP2, CDC37L1, and ECHDC2) prognostic signature could act as an independent prognostic indicator for HBV-associated HCC in both the discovery cohort and the internal testing cohort. Gene set enrichment analysis showed that the high-risk group with lower expression levels of the three genes was enriched in bladder cancer and cell cycle pathway, whereas the low-risk group with higher expression levels of the three genes was enriched in drug metabolism-cytochrome P450, PPAR signaling pathway, fatty acid and histidine metabolisms. This indicates that patients of HBV-associated HCC with higher expression of these three genes may preserve relatively good hepatic cellular metabolism and function, which may also protect HCC patients from persistent drug toxicity in response to various medication. Our findings suggest a three-gene prognostic model that serves as a specific prognostic signature for HBV-associated HCC.
Keywords: Hepatitis B virus associated hepatocellular carcinoma; Hub genes; Overall survival.; Prognostic signature; Robust Rank Aggreg analysis.
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Chibon F. Cancer gene expression signatures - the rise and fall? Eur J Cancer. 2013;49:2000–9. - PubMed
-
- Rung J, Brazma A. Reuse of public genome-wide gene expression data. Nat Rev Genet. 2013;14:89–99. - PubMed
-
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. - PubMed
-
- Zucman-Rossi J, Villanueva A, Nault J, Llovet J. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015;149:1226–39.e4. - PubMed
-
- Hoshida Y, Moeini A, Alsinet C, Kojima K, Villanueva A. Gene signatures in the management of hepatocellular carcinoma. Semin Oncol. 2012;39:473–85. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
