

Dual gain and loss of cullin 3 function mediates familial hyperkalemic hypertension
- PMID: 29897280
- PMCID: PMC6230741
- DOI: 10.1152/ajprenal.00602.2017
Dual gain and loss of cullin 3 function mediates familial hyperkalemic hypertension
Abstract
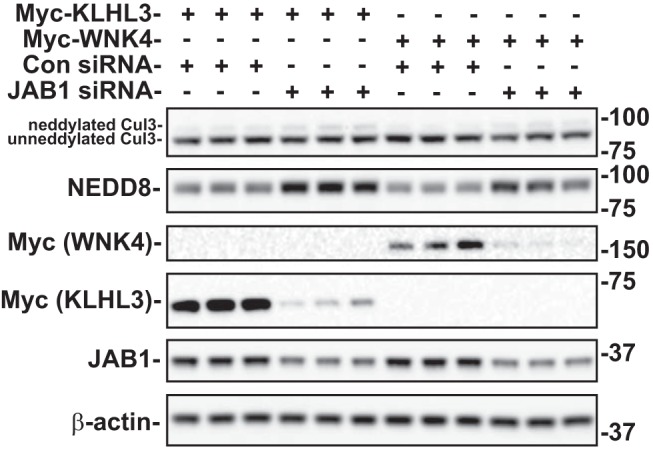
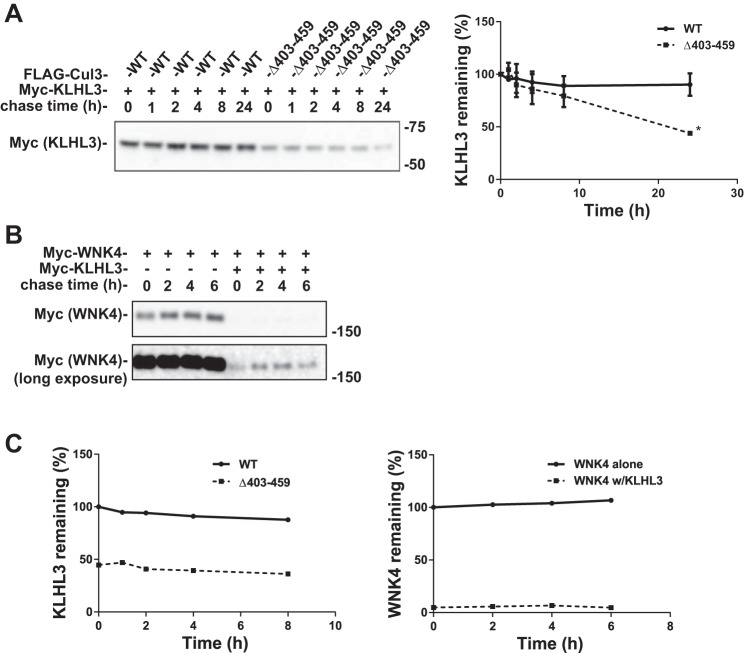
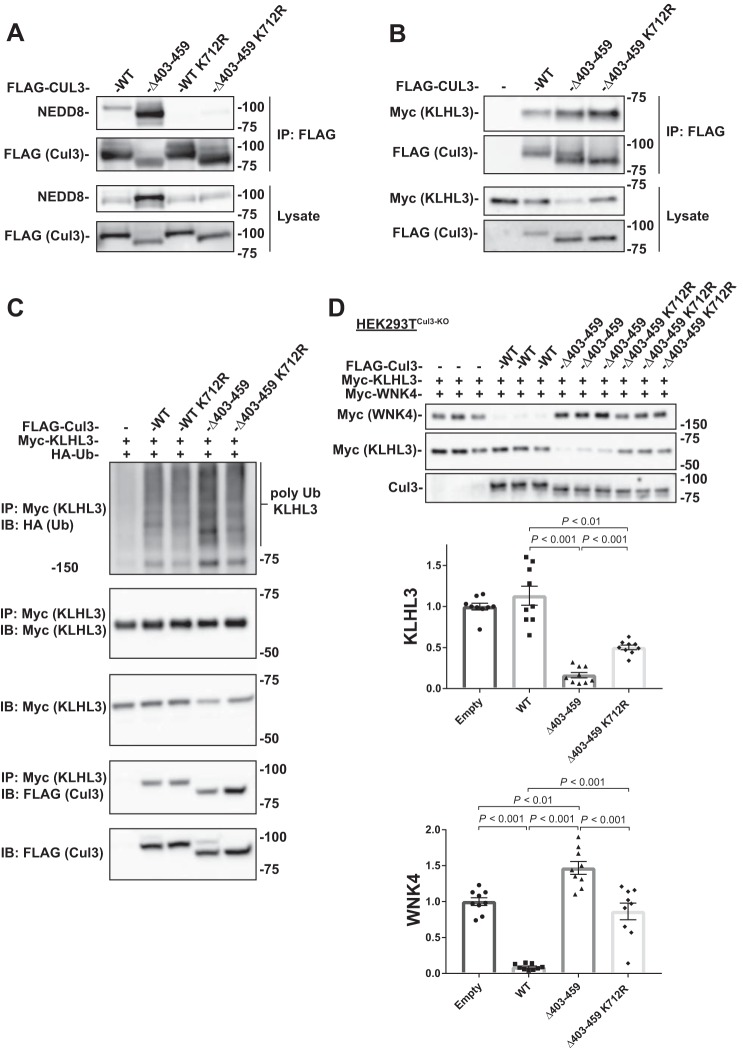
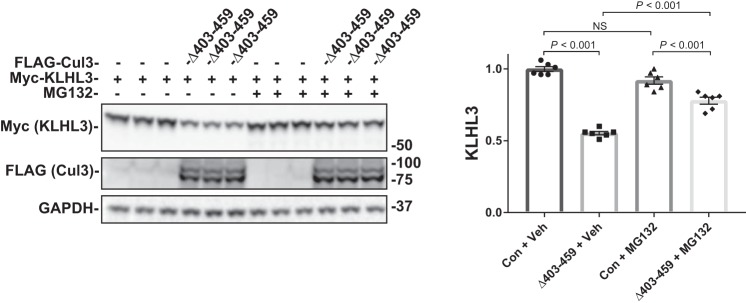
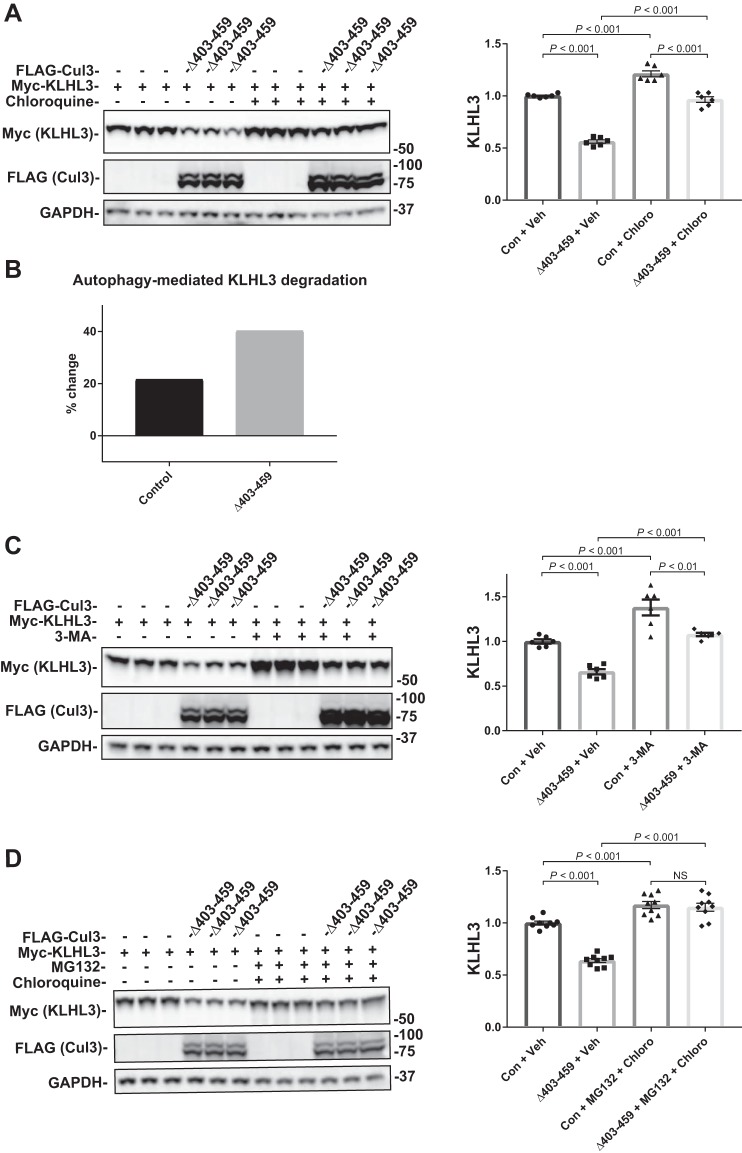
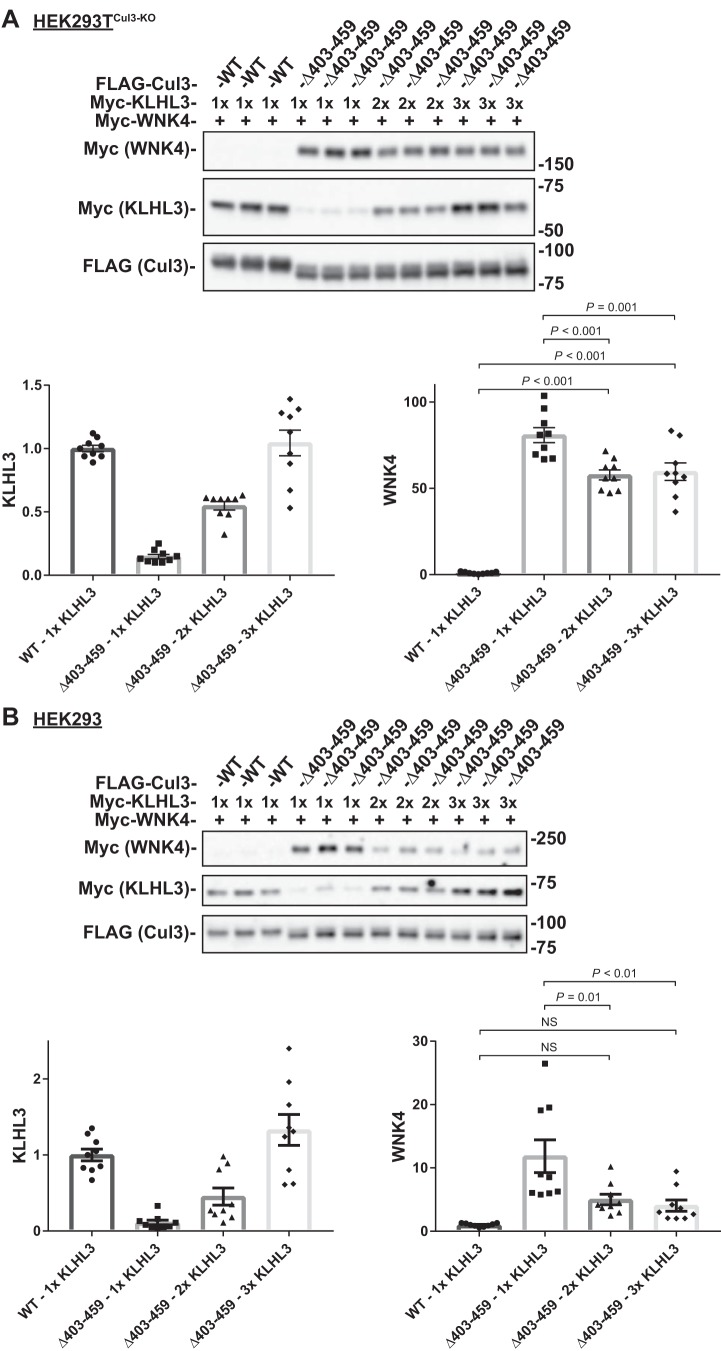
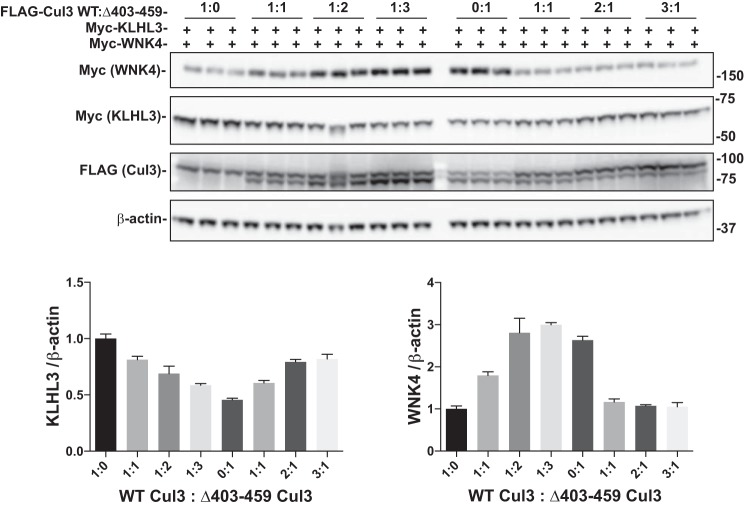
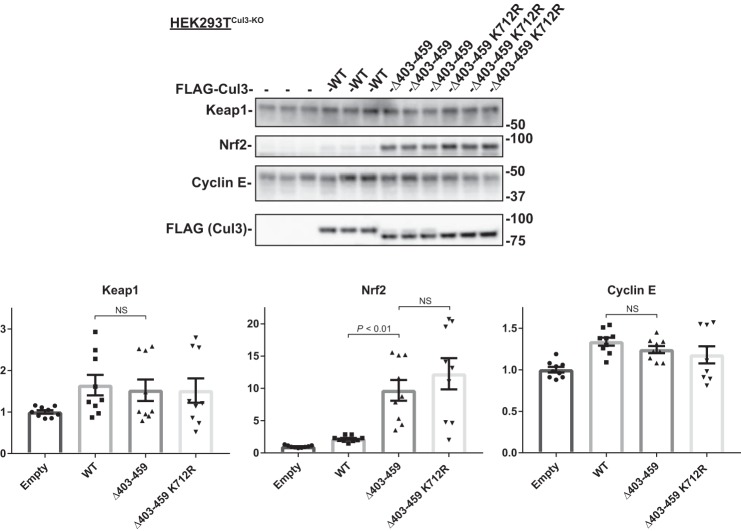
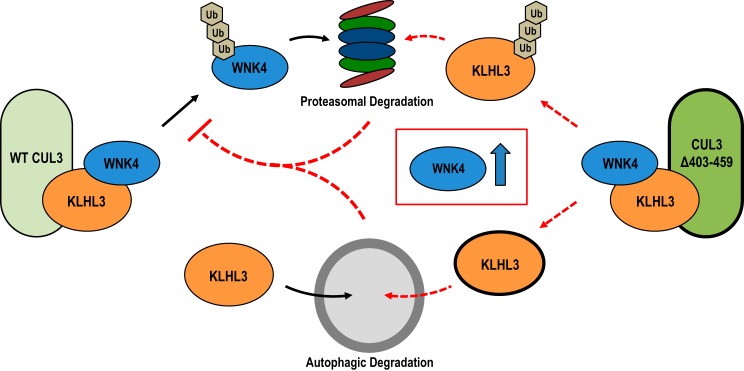
Familial hyperkalemic hypertension is caused by mutations in with-no-lysine kinases (WNKs) or in proteins that mediate their degradation, kelch-like 3 (KLHL3) and cullin 3 (CUL3). Although the mechanisms by which WNK and KLHL3 mutations cause the disease are now clear, the effects of the disease-causing CUL3Δ403-459 mutation remain controversial. Possible mechanisms, including hyperneddylation, altered ubiquitin ligase activity, decreased association with the COP9 signalosome (CSN), and increased association with and degradation of KLHL3 have all been postulated. Here, we systematically evaluated the effects of Cul3Δ403-459 using cultured kidney cells. We first identified that the catalytically active CSN subunit jun activation domain-binding protein-1 (JAB1) does not associate with the deleted Cul3 4-helix bundle domain but instead with the adjacent α/β1 domain, suggesting that altered protein folding underlies the impaired binding. Inhibition of deneddylation with JAB1 siRNA increased Cul3 neddylation and decreased KLHL3 abundance, similar to the Cul3 mutant. We next determined that KLHL3 degradation has both ubiquitin ligase-dependent and -independent components. Proteasomal KLHL3 degradation was enhanced by Cul3Δ403-459; however, autophagic degradation was also upregulated by this Cul3 mutant. Finally, to evaluate whether deficient substrate adaptor was responsible for the disease, we restored KLHL3 to wild-type (WT) Cul3 levels. In the absence of WT Cul3, WNK4 was not degraded, demonstrating that Cul3Δ403-459 itself cannot degrade WNK4; conversely, when WT Cul3 was present, as in diseased humans, WNK4 degradation was restored. In conclusion, deletion of exon 9 from Cul3 generates a protein that is itself ubiquitin-ligase defective but also capable of enhanced autophagocytic KLHL3 degradation, thereby exerting dominant-negative effects on the WT allele.
Keywords: JAB1; cullin-RING ubiquitin ligase; deneddylation; neddylation.
Figures

References
-
- Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, Lebel M, Gordon RD, Semmekrot BA, Poujol A, Välimäki MJ, De Ferrari ME, Sanjad SA, Gutkin M, Karet FE, Tucci JR, Stockigt JR, Keppler-Noreuil KM, Porter CC, Anand SK, Whiteford ML, Davis ID, Dewar SB, Bettinelli A, Fadrowski JJ, Belsha CW, Hunley TE, Nelson RD, Trachtman H, Cole TRP, Pinsk M, Bockenhauer D, Shenoy M, Vaidyanathan P, Foreman JW, Rasoulpour M, Thameem F, Al-Shahrouri HZ, Radhakrishnan J, Gharavi AG, Goilav B, Lifton RP. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482: 98–102, 2012. doi: 10.1038/nature10814. - DOI - PMC - PubMed
-
- Cavadini S, Fischer ES, Bunker RD, Potenza A, Lingaraju GM, Goldie KN, Mohamed WI, Faty M, Petzold G, Beckwith REJ, Tichkule RB, Hassiepen U, Abdulrahman W, Pantelic RS, Matsumoto S, Sugasawa K, Stahlberg H, Thomä NH. Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 531: 598–603, 2016. doi: 10.1038/nature17416. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous