

CONICS integrates scRNA-seq with DNA sequencing to map gene expression to tumor sub-clones
- PMID: 29897414
- PMCID: PMC7190654
- DOI: 10.1093/bioinformatics/bty316
CONICS integrates scRNA-seq with DNA sequencing to map gene expression to tumor sub-clones
Abstract
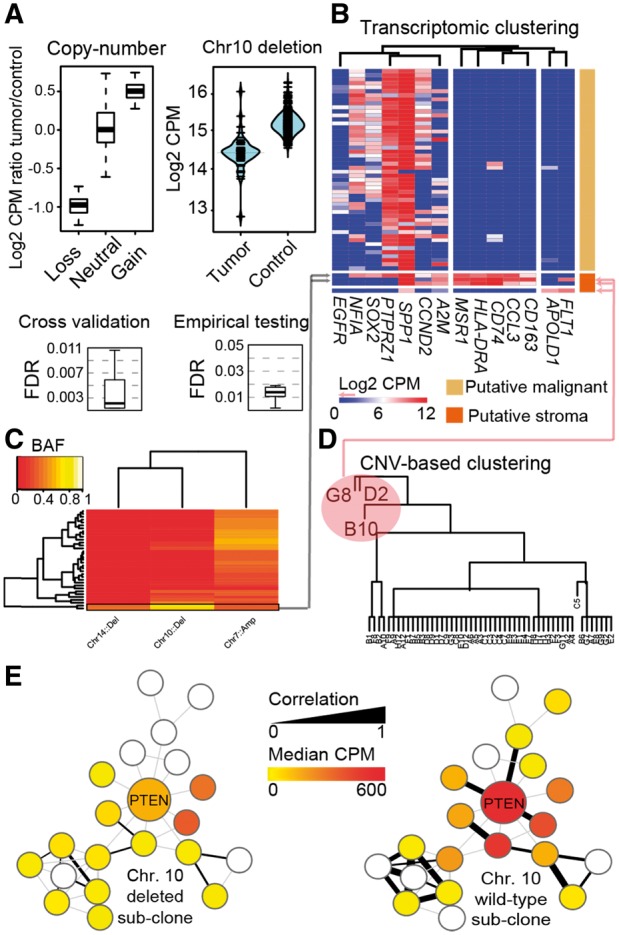
Motivation: Single-cell RNA-sequencing (scRNA-seq) has enabled studies of tissue composition at unprecedented resolution. However, the application of scRNA-seq to clinical cancer samples has been limited, partly due to a lack of scRNA-seq algorithms that integrate genomic mutation data.
Results: To address this, we present.
Conics: COpy-Number analysis In single-Cell RNA-Sequencing. CONICS is a software tool for mapping gene expression from scRNA-seq to tumor clones and phylogenies, with routines enabling: the quantitation of copy-number alterations in scRNA-seq, robust separation of neoplastic cells from tumor-infiltrating stroma, inter-clone differential-expression analysis and intra-clone co-expression analysis.
Availability and implementation: CONICS is written in Python and R, and is available from https://github.com/diazlab/CONICS.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
