

Pre-clinical symptoms of SBMA may not be androgen-dependent: implications from two SBMA mouse models
- PMID: 29897452
- PMCID: PMC6030880
- DOI: 10.1093/hmg/ddy142
Pre-clinical symptoms of SBMA may not be androgen-dependent: implications from two SBMA mouse models
Abstract
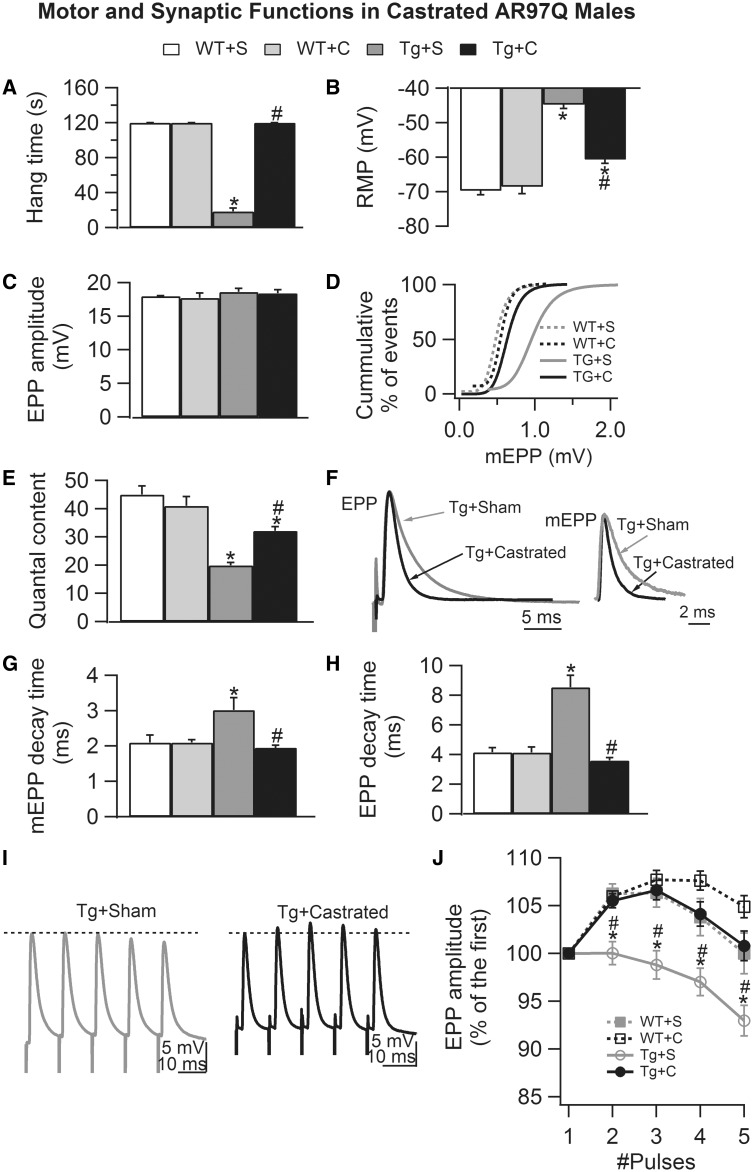
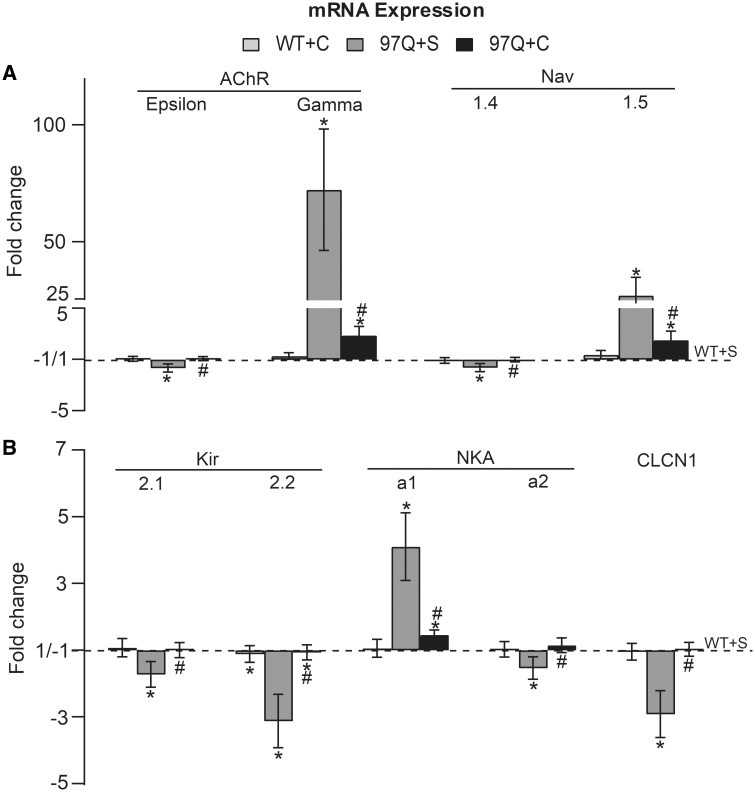
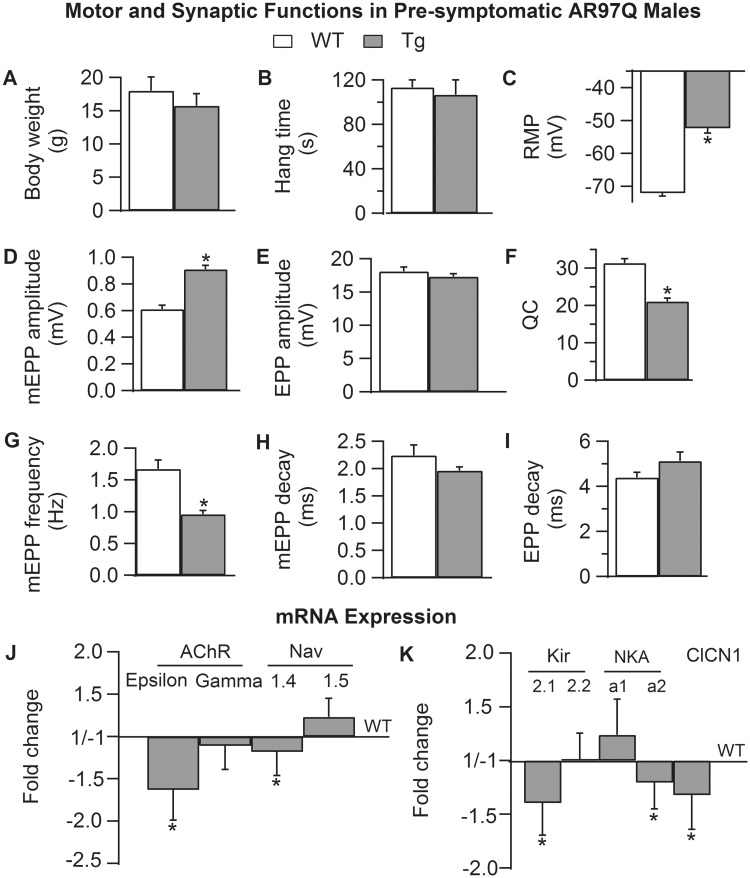
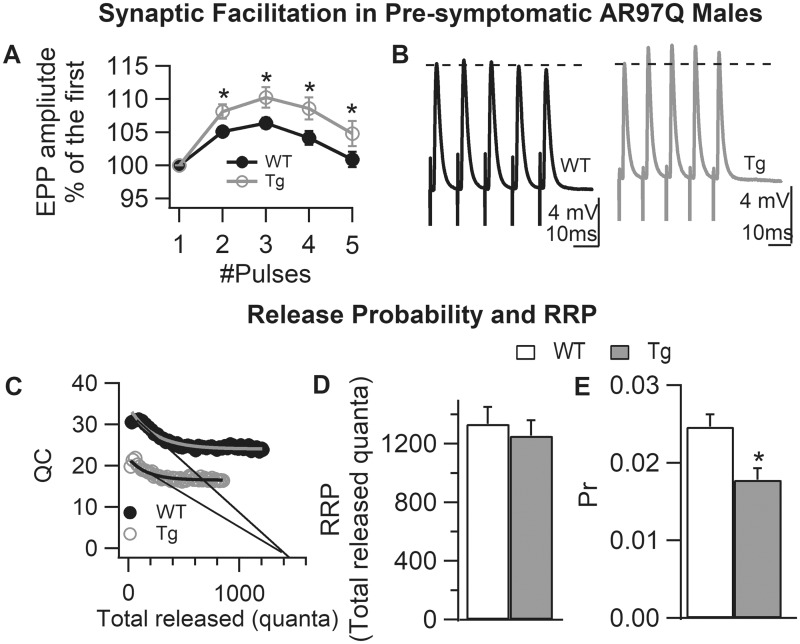
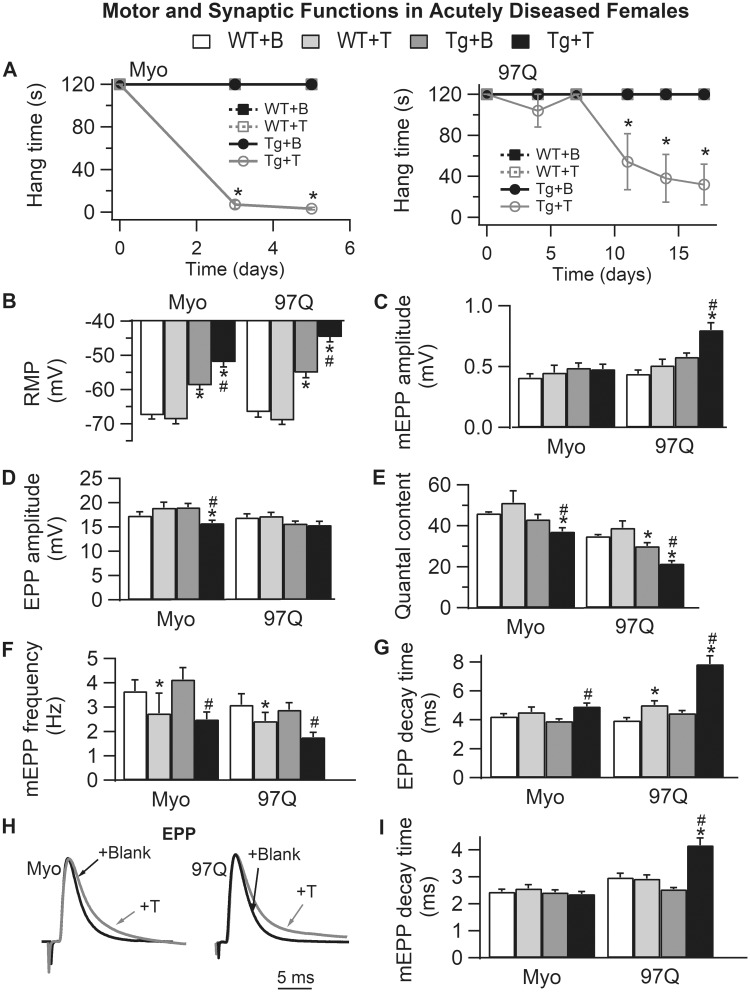
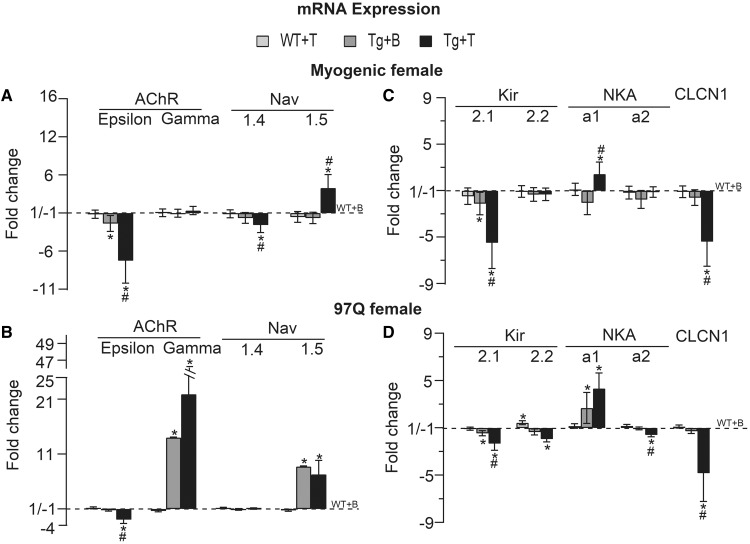
A distinguishing aspect of spinal and bulbar muscular atrophy (SBMA) is its androgen-dependence, possibly explaining why only males are clinically affected. This disease, which impairs neuromuscular function, is linked to a polyglutamine expansion mutation in the androgen receptor (AR). In mouse models of SBMA, motor dysfunction is associated with pronounced defects in neuromuscular transmission, including defects in evoked transmitter release (quantal content, QC) and fiber membrane excitability (based on the resting membrane potential, RMP). However, whether such defects are androgen-dependent is unknown. Thus, we recorded synaptic potentials intracellularly from adult muscle fibers of transgenic (Tg) AR97Q male mice castrated pre-symptomatically. Although castration largely protects both QC and the RMP of fibers, correlating with the protective effect of castration on motor function, significant deficits in QC and RMP remained. Surprisingly, comparable defects in QC and RMP were also observed in pre-symptomatic AR97Q males, indicating that such defects emerge early and are pre-clinical. Exposing asymptomatic Tg females to androgens also induces both motor dysfunction and comparable defects in QC and RMP. Notably, asymptomatic Tg females also showed significant deficits in QC and RMP, albeit less severe, supporting their pre-clinical nature, but also raising questions about the androgen-dependence of pre-clinical symptoms. In summary, current evidence indicates that disease progression depends on androgens, but early pathogenic events may be triggered by the mutant AR allele independent of androgens. Such early, androgen-independent disease mechanisms may also be relevant to females carrying the SBMA allele.
Figures
Similar articles
-
Antiandrogen flutamide protects male mice from androgen-dependent toxicity in three models of spinal bulbar muscular atrophy.Endocrinology. 2014 Jul;155(7):2624-34. doi: 10.1210/en.2013-1756. Epub 2014 Apr 17. Endocrinology. 2014. PMID: 24742193 Free PMC article.
-
Recovery of function in a myogenic mouse model of spinal bulbar muscular atrophy.Neurobiol Dis. 2009 Apr;34(1):113-20. doi: 10.1016/j.nbd.2008.12.009. Epub 2008 Dec 31. Neurobiol Dis. 2009. PMID: 19211034 Free PMC article.
-
Defects in Neuromuscular Transmission May Underlie Motor Dysfunction in Spinal and Bulbar Muscular Atrophy.J Neurosci. 2016 May 4;36(18):5094-106. doi: 10.1523/JNEUROSCI.3485-15.2016. J Neurosci. 2016. PMID: 27147661 Free PMC article.
-
X-Linked Spinal and Bulbar Muscular Atrophy: From Clinical Genetic Features and Molecular Pathology to Mechanisms Underlying Disease Toxicity.Adv Exp Med Biol. 2018;1049:103-133. doi: 10.1007/978-3-319-71779-1_5. Adv Exp Med Biol. 2018. PMID: 29427100 Review.
-
In Vitro and In Vivo Modeling of Spinal and Bulbar Muscular Atrophy.J Mol Neurosci. 2016 Mar;58(3):365-73. doi: 10.1007/s12031-015-0677-4. Epub 2015 Nov 27. J Mol Neurosci. 2016. PMID: 26614347 Review.
Cited by
-
Autophagic and Proteasomal Mediated Removal of Mutant Androgen Receptor in Muscle Models of Spinal and Bulbar Muscular Atrophy.Front Endocrinol (Lausanne). 2019 Aug 20;10:569. doi: 10.3389/fendo.2019.00569. eCollection 2019. Front Endocrinol (Lausanne). 2019. PMID: 31481932 Free PMC article.
-
Neuromuscular junction pathology is correlated with differential motor unit vulnerability in spinal and bulbar muscular atrophy.Acta Neuropathol Commun. 2022 Jul 5;10(1):97. doi: 10.1186/s40478-022-01402-y. Acta Neuropathol Commun. 2022. PMID: 35791011 Free PMC article.
-
Molecular Mechanisms and Therapeutics for SBMA/Kennedy's Disease.Neurotherapeutics. 2019 Oct;16(4):928-947. doi: 10.1007/s13311-019-00790-9. Neurotherapeutics. 2019. PMID: 31686397 Free PMC article. Review.
-
The Regulation of the Small Heat Shock Protein B8 in Misfolding Protein Diseases Causing Motoneuronal and Muscle Cell Death.Front Neurosci. 2019 Aug 2;13:796. doi: 10.3389/fnins.2019.00796. eCollection 2019. Front Neurosci. 2019. PMID: 31427919 Free PMC article. Review.
-
Disease Affects Bdnf Expression in Synaptic and Extrasynaptic Regions of Skeletal Muscle of Three SBMA Mouse Models.Int J Mol Sci. 2019 Mar 15;20(6):1314. doi: 10.3390/ijms20061314. Int J Mol Sci. 2019. PMID: 30875922 Free PMC article.
References
-
- Sobue G., Hashizume Y., Mukai E., Hirayama M., Mitsuma T., Takahashi A. (1989) X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain, 112, 209–232. - PubMed
-
- La Spada A.R., Wilson E.M., Lubahn D.B., Harding A.E., Fischbeck K.H. (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature, 352, 77–79. - PubMed
-
- Schmidt B.J., Greenberg C.R., Allingham-Hawkins D.J., Spriggs E.L. (2002) Expression of X-linked bulbospinal muscular atrophy (Kennedy disease) in two homozygous women. Neurology, 59, 770–772. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
