

Deducing the presence of proteins and proteoforms in quantitative proteomics
- PMID: 29899466
- PMCID: PMC5998138
- DOI: 10.1038/s41467-018-04411-5
Deducing the presence of proteins and proteoforms in quantitative proteomics
Abstract
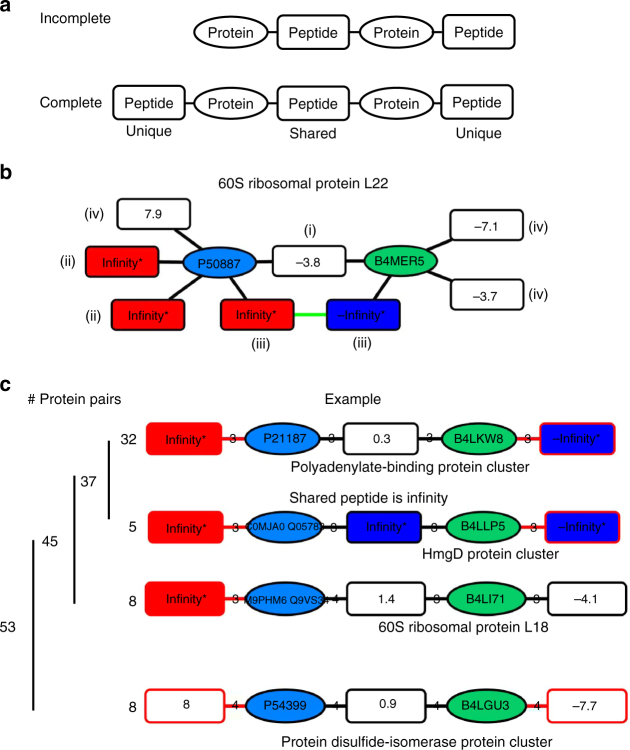
The human genome harbors just 20,000 genes suggesting that the variety of possible protein products per gene plays a significant role in generating functional diversity. In bottom-up proteomics peptides are mapped back to proteins and proteoforms to describe a proteome; however, accurate quantitation of proteoforms is challenging due to incomplete protein sequence coverage and mapping ambiguities. Here, we demonstrate that a new software tool called ProteinClusterQuant (PCQ) can be used to deduce the presence of proteoforms that would have otherwise been missed, as exemplified in a proteomic comparison of two fly species, Drosophila melanogaster and D. virilis. PCQ was used to identify reduced levels of serine/threonine protein kinases PKN1 and PKN4 in CFBE41o- cells compared to HBE41o- cells and to elucidate that shorter proteoforms of full-length caspase-4 and ephrin B receptor are differentially expressed. Thus, PCQ extends current analyses in quantitative proteomics and facilitates finding differentially regulated proteins and proteoforms.
Conflict of interest statement
The authors declare no competing interests.
Figures






Similar articles
-
A high-quality catalog of the Drosophila melanogaster proteome.Nat Biotechnol. 2007 May;25(5):576-83. doi: 10.1038/nbt1300. Epub 2007 Apr 22. Nat Biotechnol. 2007. PMID: 17450130
-
The Drosophila melanogaster PeptideAtlas facilitates the use of peptide data for improved fly proteomics and genome annotation.BMC Bioinformatics. 2009 Feb 11;10:59. doi: 10.1186/1471-2105-10-59. BMC Bioinformatics. 2009. PMID: 19210778 Free PMC article.
-
Proteomics of the Drosophila immune response.Trends Biotechnol. 2004 Nov;22(11):600-5. doi: 10.1016/j.tibtech.2004.09.002. Trends Biotechnol. 2004. PMID: 15491805 Review.
-
A mass graph-based approach for the identification of modified proteoforms using top-down tandem mass spectra.Bioinformatics. 2017 May 1;33(9):1309-1316. doi: 10.1093/bioinformatics/btw806. Bioinformatics. 2017. PMID: 28453668 Free PMC article.
-
Profiling proteoforms: promising follow-up of proteomics for biomarker discovery.Expert Rev Proteomics. 2014 Feb;11(1):121-9. doi: 10.1586/14789450.2014.878652. Expert Rev Proteomics. 2014. PMID: 24437377 Review.
Cited by
-
Functional proteoform group deconvolution reveals a broader spectrum of ibrutinib off-targets.Nat Commun. 2025 Feb 25;16(1):1948. doi: 10.1038/s41467-024-54654-8. Nat Commun. 2025. PMID: 40000607 Free PMC article.
-
Cancer Conformational Landscape Shapes Tumorigenesis.J Proteome Res. 2022 Apr 1;21(4):1017-1028. doi: 10.1021/acs.jproteome.1c00906. Epub 2022 Mar 10. J Proteome Res. 2022. PMID: 35271278 Free PMC article.
-
Quantitative structural proteomics in living cells by covalent protein painting.Methods Enzymol. 2023;679:33-63. doi: 10.1016/bs.mie.2022.08.046. Epub 2023 Jan 4. Methods Enzymol. 2023. PMID: 36682868 Free PMC article.
-
Characterization of peptide-protein relationships in protein ambiguity groups via bipartite graphs.PLoS One. 2022 Oct 21;17(10):e0276401. doi: 10.1371/journal.pone.0276401. eCollection 2022. PLoS One. 2022. PMID: 36269744 Free PMC article.
-
Putting Humpty Dumpty Back Together Again: What Does Protein Quantification Mean in Bottom-Up Proteomics?J Proteome Res. 2022 Apr 1;21(4):891-898. doi: 10.1021/acs.jproteome.1c00894. Epub 2022 Feb 27. J Proteome Res. 2022. PMID: 35220718 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- P41 GM103533/GM/NIGMS NIH HHS/United States
- P01AG031097/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)/International
- R33 CA212973/CA/NCI NIH HHS/United States
- P01 AG031097/AG/NIA NIH HHS/United States
- P41 GM103533/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
