

Long-read sequencing of nascent RNA reveals coupling among RNA processing events
- PMID: 29903723
- PMCID: PMC6028129
- DOI: 10.1101/gr.232025.117
Long-read sequencing of nascent RNA reveals coupling among RNA processing events
Abstract
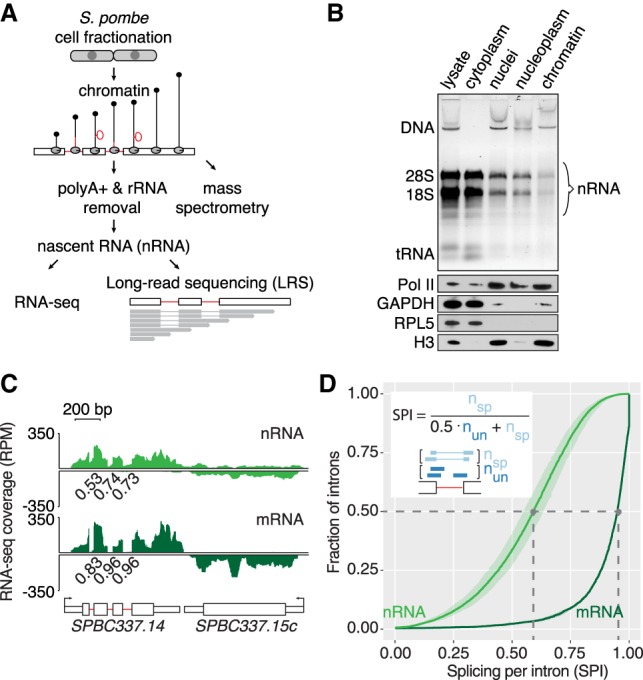
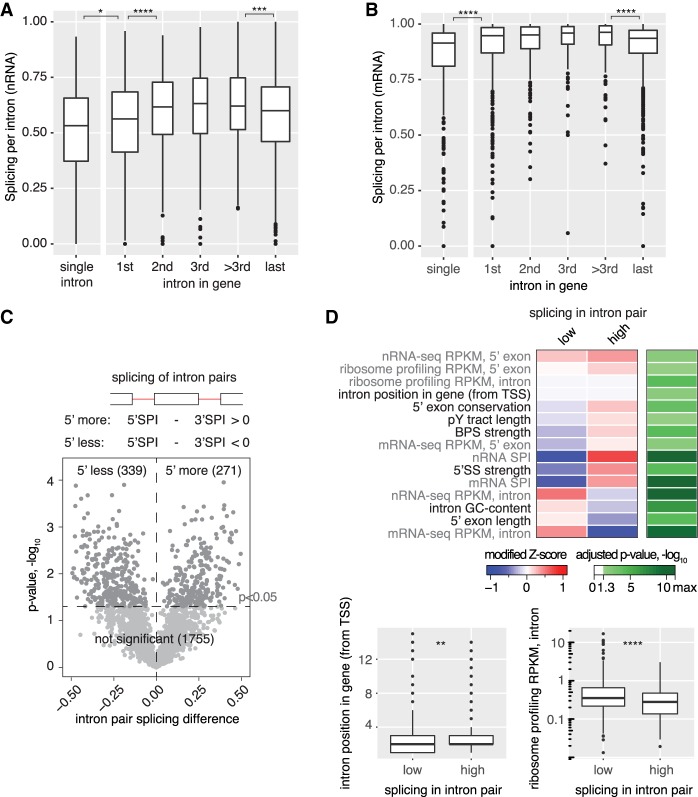
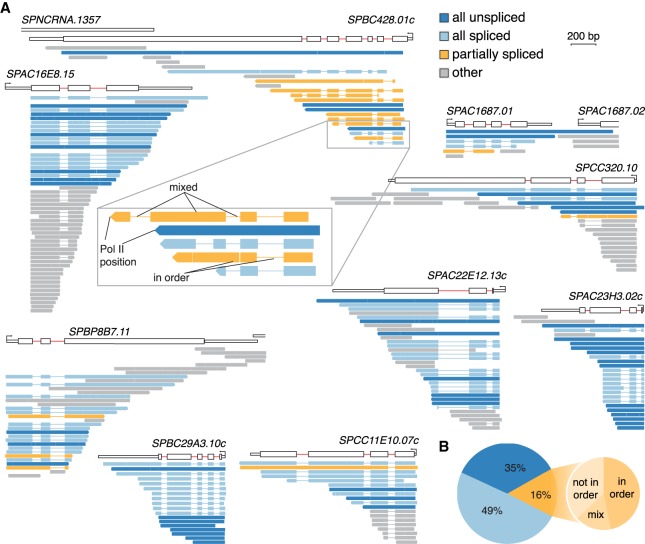
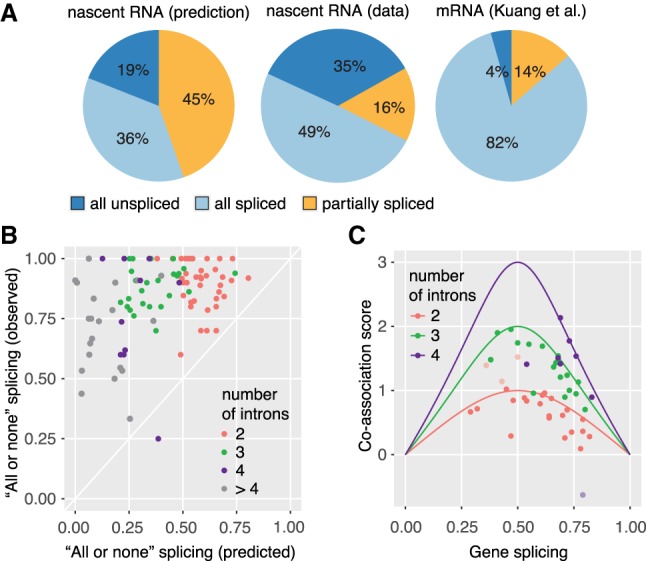
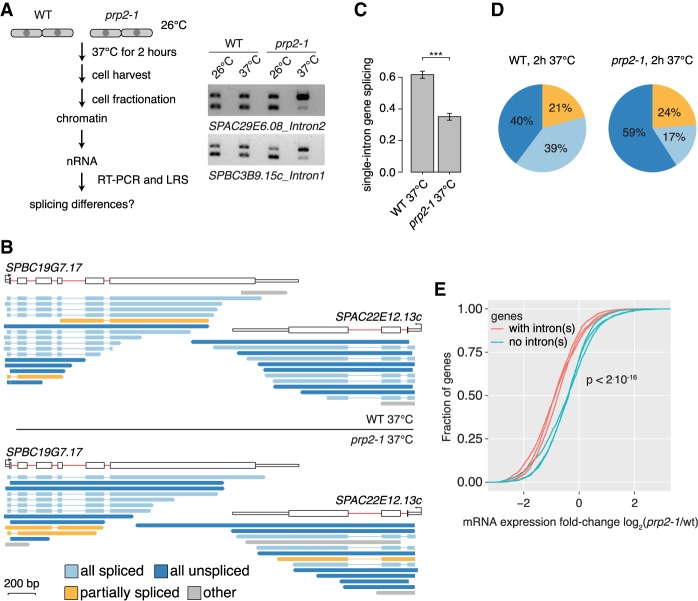
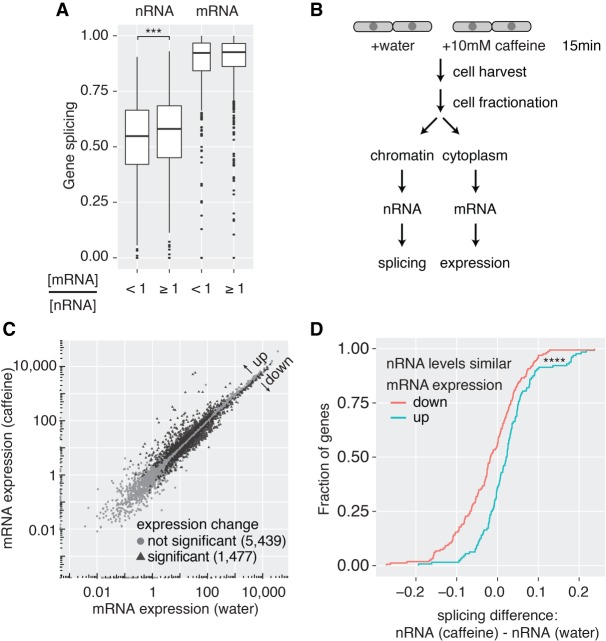
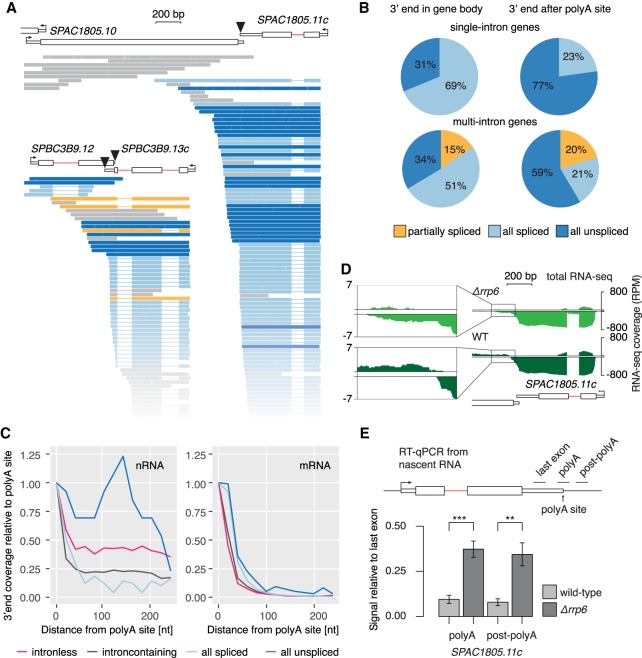
Pre-mRNA splicing is accomplished by the spliceosome, a megadalton complex that assembles de novo on each intron. Because spliceosome assembly and catalysis occur cotranscriptionally, we hypothesized that introns are removed in the order of their transcription in genomes dominated by constitutive splicing. Remarkably little is known about splicing order and the regulatory potential of nascent transcript remodeling by splicing, due to the limitations of existing methods that focus on analysis of mature splicing products (mRNAs) rather than substrates and intermediates. Here, we overcome this obstacle through long-read RNA sequencing of nascent, multi-intron transcripts in the fission yeast Schizosaccharomyces pombe Most multi-intron transcripts were fully spliced, consistent with rapid cotranscriptional splicing. However, an unexpectedly high proportion of transcripts were either fully spliced or fully unspliced, suggesting that splicing of any given intron is dependent on the splicing status of other introns in the transcript. Supporting this, mild inhibition of splicing by a temperature-sensitive mutation in prp2, the homolog of vertebrate U2AF65, increased the frequency of fully unspliced transcripts. Importantly, fully unspliced transcripts displayed transcriptional read-through at the polyA site and were degraded cotranscriptionally by the nuclear exosome. Finally, we show that cellular mRNA levels were reduced in genes with a high number of unspliced nascent transcripts during caffeine treatment, showing regulatory significance of cotranscriptional splicing. Therefore, overall splicing of individual nascent transcripts, 3' end formation, and mRNA half-life depend on the splicing status of neighboring introns, suggesting crosstalk among spliceosomes and the polyA cleavage machinery during transcription elongation.
© 2018 Herzel et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Alexander RD, Barrass JD, Dichtl B, Kos M, Obtulowicz T, Robert MC, Koper M, Karkusiewicz I, Mariconti L, Tollervey D, et al. 2010. RiboSys, a high-resolution, quantitative approach to measure the in vivo kinetics of pre-mRNA splicing and 3′-end processing in Saccharomyces cerevisiae. RNA 16: 2570–2580. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases