

Diazepam-induced loss of inhibitory synapses mediated by PLCδ/ Ca2+/calcineurin signalling downstream of GABAA receptors
- PMID: 29904150
- PMCID: PMC6232101
- DOI: 10.1038/s41380-018-0100-y
Diazepam-induced loss of inhibitory synapses mediated by PLCδ/ Ca2+/calcineurin signalling downstream of GABAA receptors
Abstract
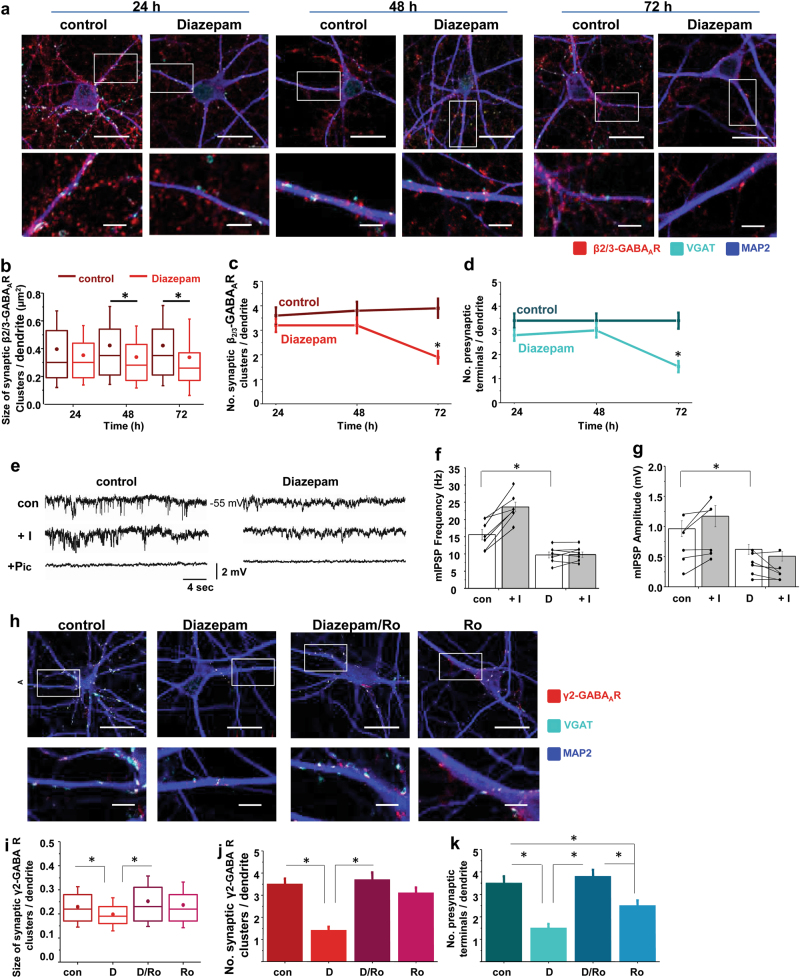
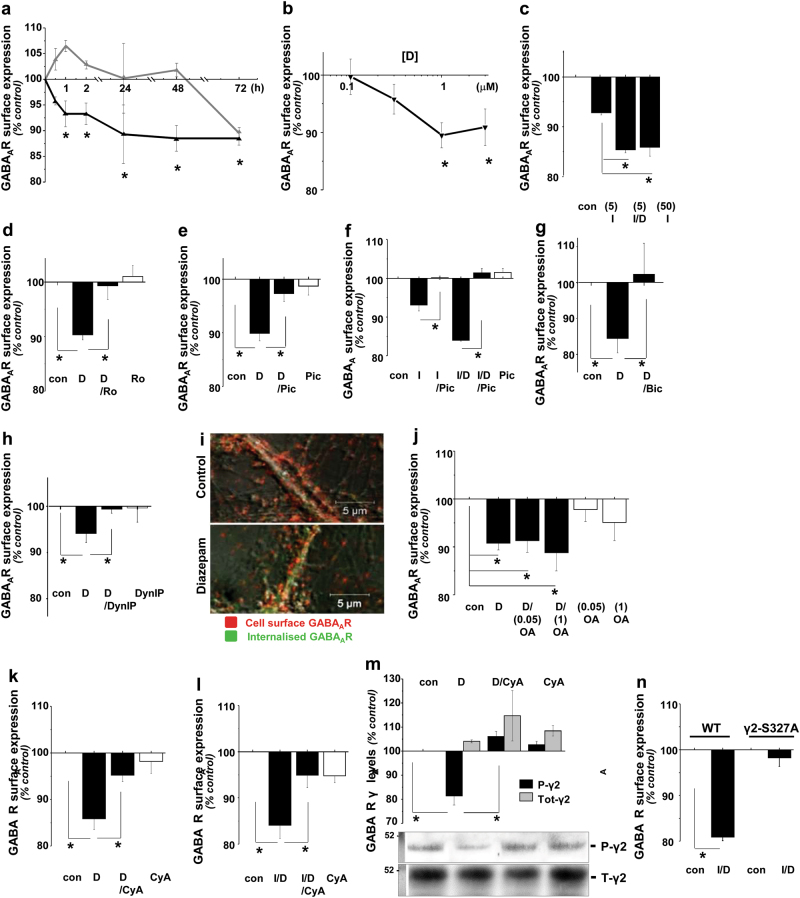
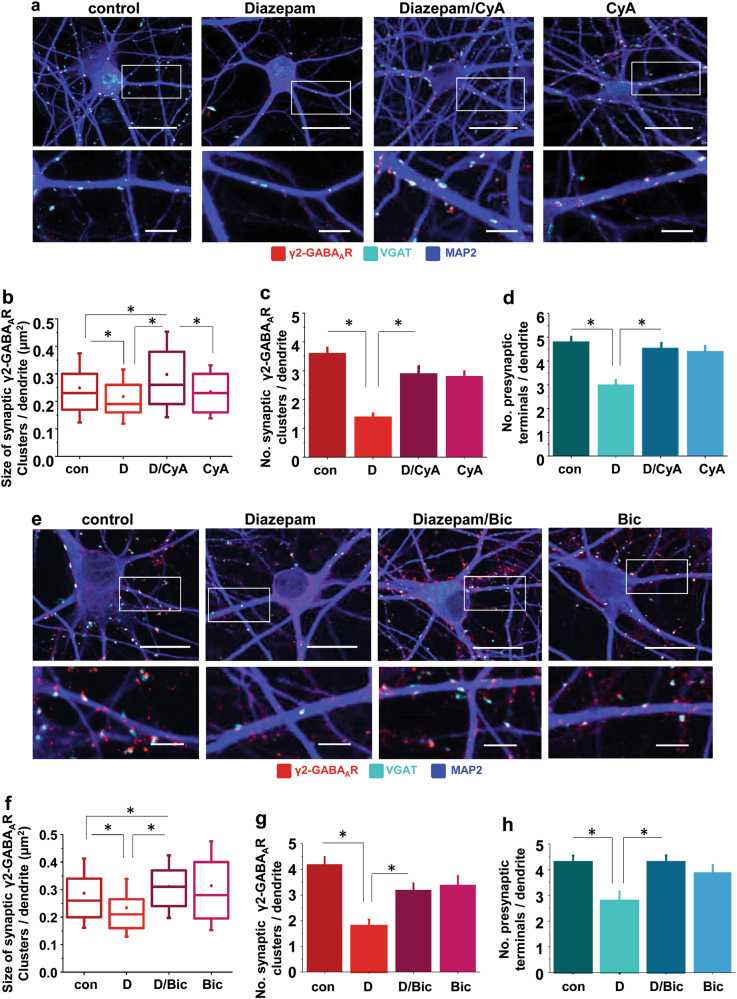
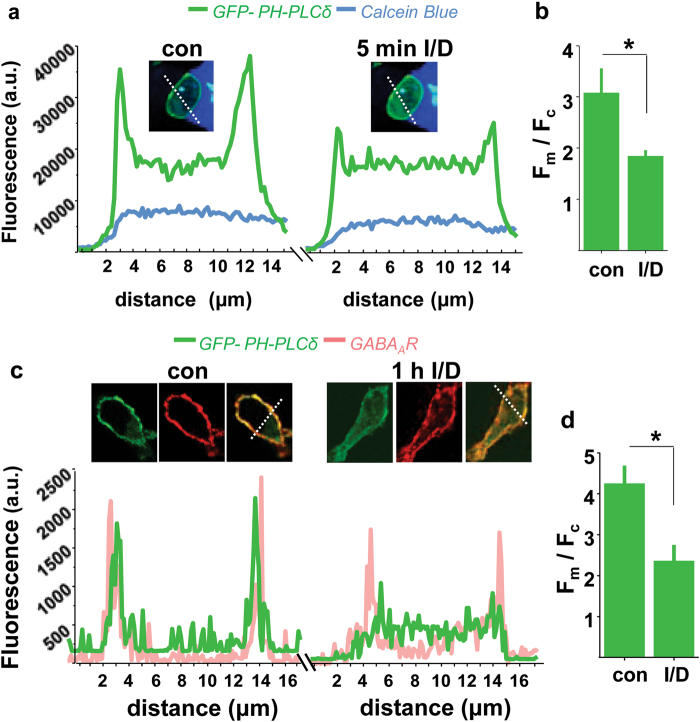
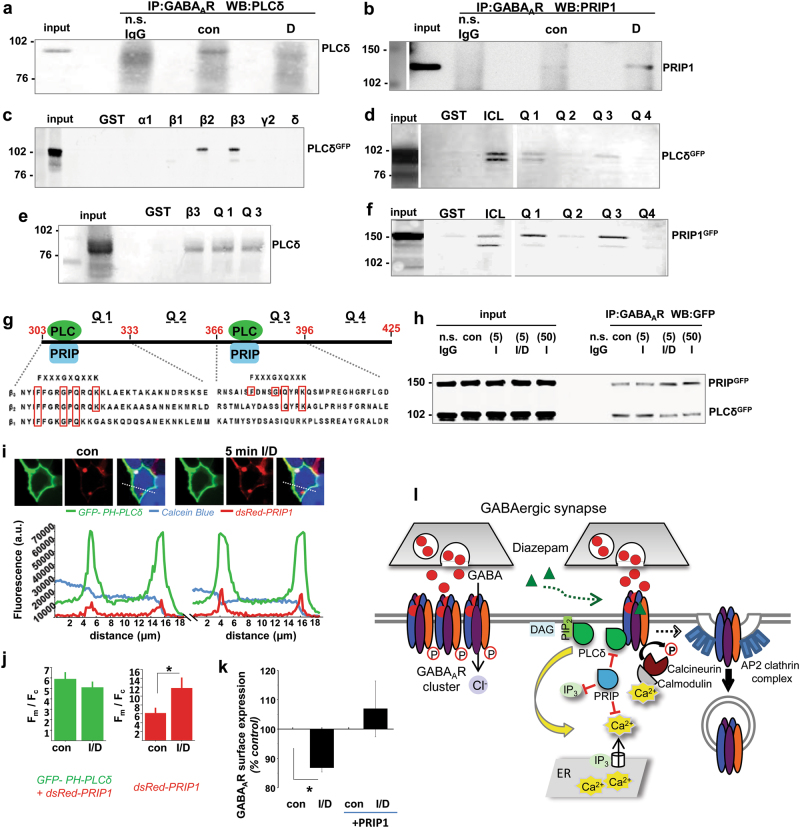
Benzodiazepines facilitate the inhibitory actions of GABA by binding to γ-aminobutyric acid type A receptors (GABAARs), GABA-gated chloride/bicarbonate channels, which are the key mediators of transmission at inhibitory synapses in the brain. This activity underpins potent anxiolytic, anticonvulsant and hypnotic effects of benzodiazepines in patients. However, extended benzodiazepine treatments lead to development of tolerance, a process which, despite its important therapeutic implications, remains poorly characterised. Here we report that prolonged exposure to diazepam, the most widely used benzodiazepine in clinic, leads to a gradual disruption of neuronal inhibitory GABAergic synapses. The loss of synapses and the preceding, time- and dose-dependent decrease in surface levels of GABAARs, mediated by dynamin-dependent internalisation, were blocked by Ro 15-1788, a competitive benzodiazepine antagonist, and bicuculline, a competitive GABA antagonist, indicating that prolonged enhancement of GABAAR activity by diazepam is integral to the underlying molecular mechanism. Characterisation of this mechanism has revealed a metabotropic-type signalling downstream of GABAARs, involving mobilisation of Ca2+ from the intracellular stores and activation of the Ca2+/calmodulin-dependent phosphatase calcineurin, which, in turn, dephosphorylates GABAARs and promotes their endocytosis, leading to disassembly of inhibitory synapses. Furthermore, functional coupling between GABAARs and Ca2+ stores was sensitive to phospholipase C (PLC) inhibition by U73122, and regulated by PLCδ, a PLC isoform found in direct association with GABAARs. Thus, a PLCδ/Ca2+/calcineurin signalling cascade converts the initial enhancement of GABAARs by benzodiazepines to a long-term downregulation of GABAergic synapses, this potentially underpinning the development of pharmacological and behavioural tolerance to these widely prescribed drugs.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
