

GFPT1 deficiency in muscle leads to myasthenia and myopathy in mice
- PMID: 29905857
- PMCID: PMC6121184
- DOI: 10.1093/hmg/ddy225
GFPT1 deficiency in muscle leads to myasthenia and myopathy in mice
Abstract
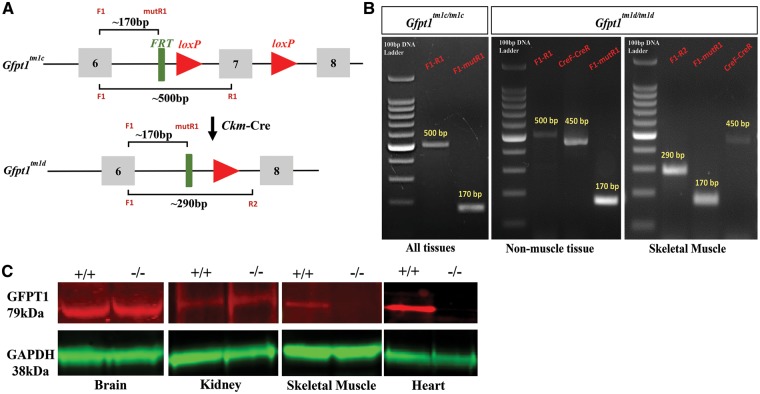
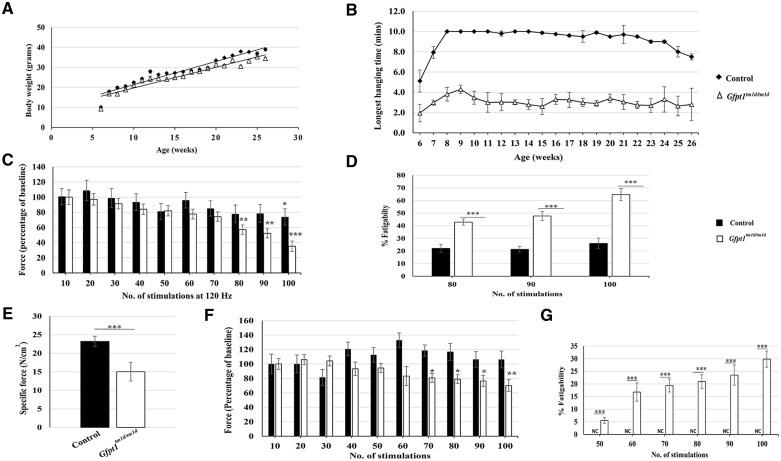
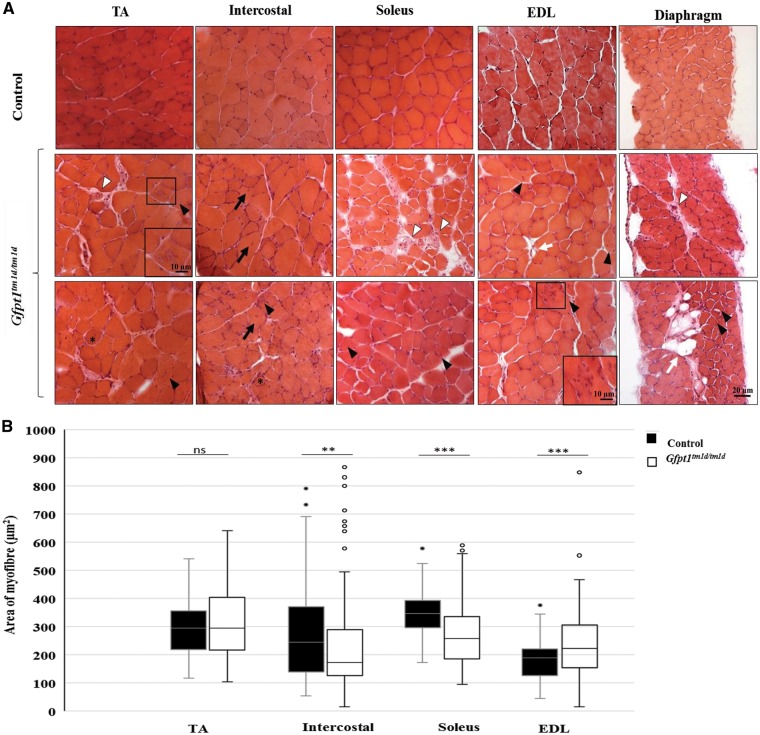
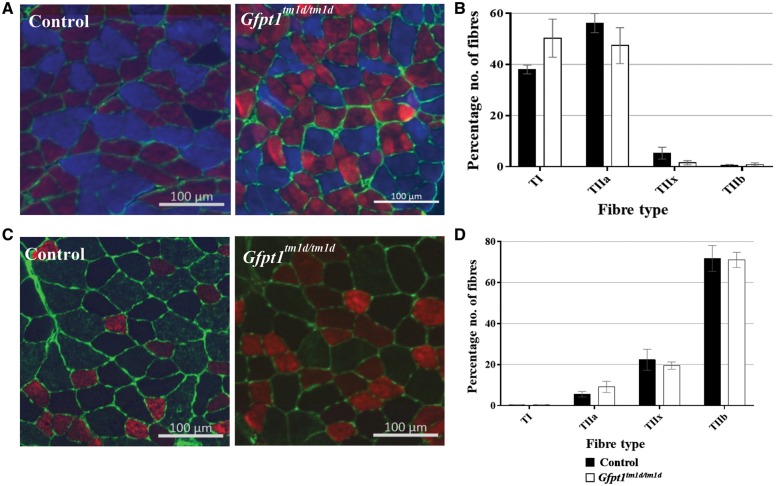
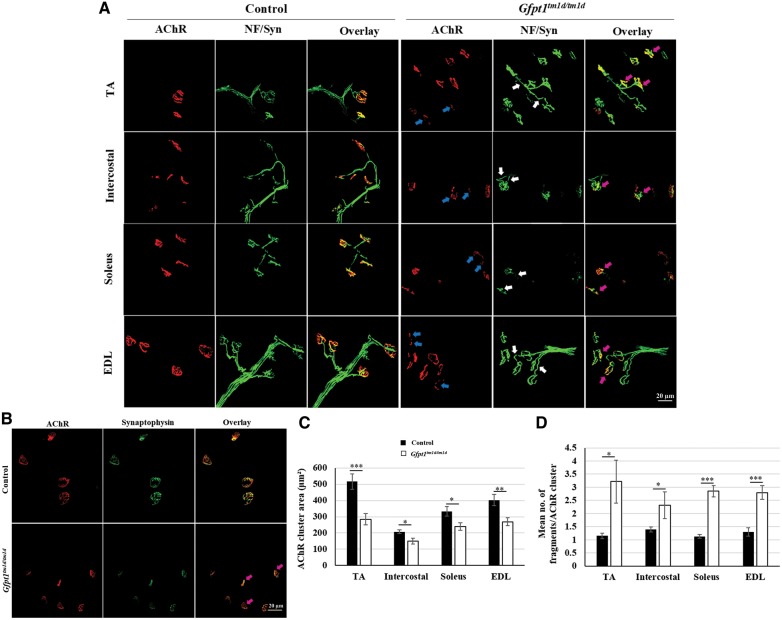
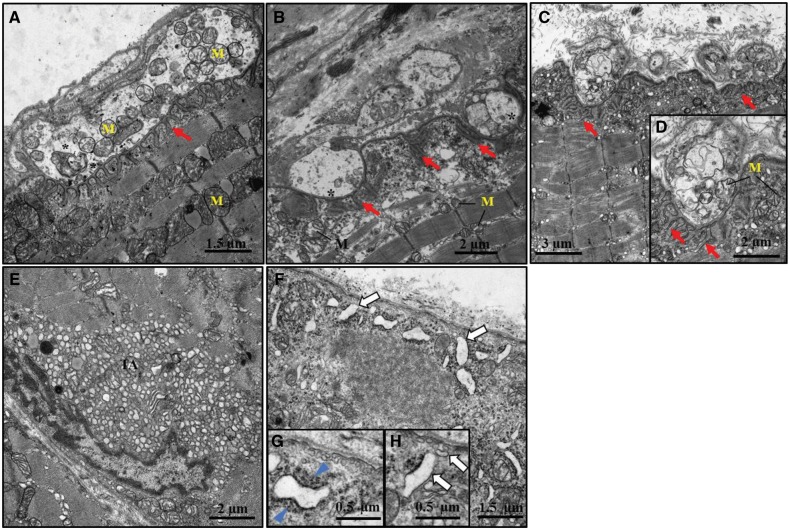
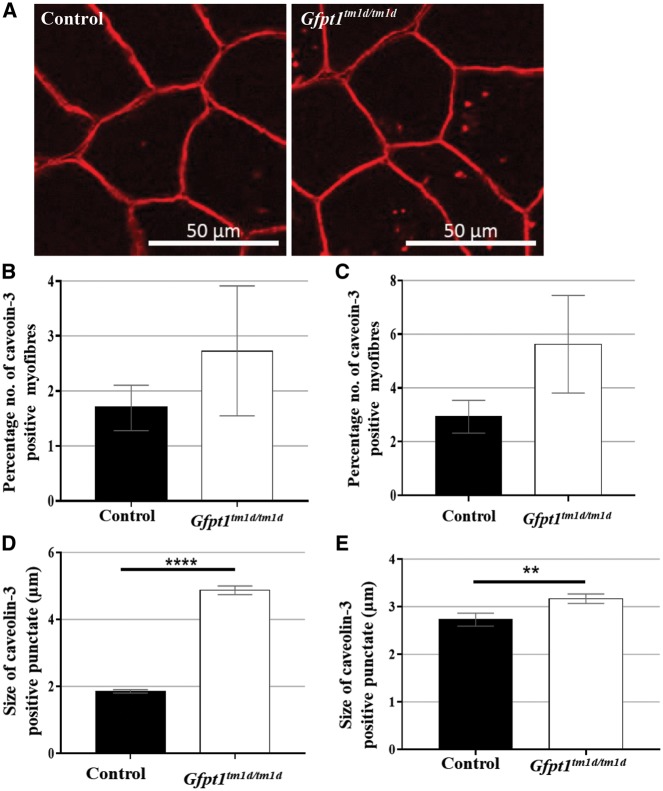
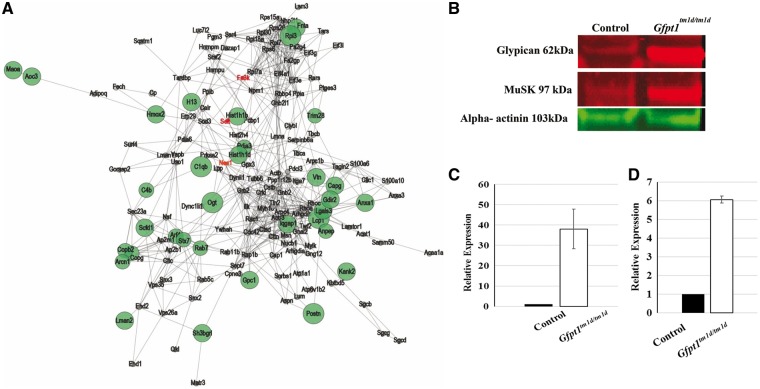
Glutamine-fructose-6-phosphate transaminase 1 (GFPT1) is the rate-limiting enzyme in the hexosamine biosynthetic pathway which yields precursors required for protein and lipid glycosylation. Mutations in GFPT1 and other genes downstream of this pathway cause congenital myasthenic syndrome (CMS) characterized by fatigable muscle weakness owing to impaired neurotransmission. The precise pathomechanisms at the neuromuscular junction (NMJ) owing to a deficiency in GFPT1 is yet to be discovered. One of the challenges we face is the viability of Gfpt1-/- knockout mice. In this study, we use Cre/LoxP technology to generate a muscle-specific GFPT1 knockout mouse model, Gfpt1tm1d/tm1d, characteristic of the human CMS phenotype. Our data suggest a critical role for muscle derived GFPT1 in the development of the NMJ, neurotransmission, skeletal muscle integrity and highlight that a deficiency in skeletal muscle alone is sufficient to cause morphological postsynaptic NMJ changes that are accompanied by presynaptic alterations despite the conservation of neuronal GFPT1 expression. In addition to the conventional morphological NMJ changes and fatigable muscle weakness, Gfpt1tm1d/tm1d mice display a progressive myopathic phenotype with the presence of tubular aggregates in muscle, characteristic of the GFPT1-CMS phenotype. We further identify an upregulation of skeletal muscle proteins glypican-1, farnesyltransferase/geranylgeranyltransferase type-1 subunit α and muscle-specific kinase, which are known to be involved in the differentiation and maintenance of the NMJ. The Gfpt1tm1d/tm1d model allows for further investigation of pathophysiological consequences on genes and pathways downstream of GFPT1 likely to involve misglycosylation or hypoglycosylation of NMJs and muscle targets.
Figures
Similar articles
-
A Deficiency in Glutamine-Fructose-6-Phosphate Transaminase 1 (Gfpt1) in Skeletal Muscle Results in Reduced Glycosylation of the Delta Subunit of the Nicotinic Acetylcholine Receptor (AChRδ).Biomolecules. 2024 Oct 3;14(10):1252. doi: 10.3390/biom14101252. Biomolecules. 2024. PMID: 39456185 Free PMC article.
-
Mutations in GFPT1-related congenital myasthenic syndromes are associated with synaptic morphological defects and underlie a tubular aggregate myopathy with synaptopathy.J Neurol. 2017 Aug;264(8):1791-1803. doi: 10.1007/s00415-017-8569-x. Epub 2017 Jul 15. J Neurol. 2017. PMID: 28712002
-
Diverse myopathological features in the congenital myasthenia syndrome with GFPT1 mutation.Brain Behav. 2022 Feb;12(2):e2469. doi: 10.1002/brb3.2469. Epub 2022 Jan 3. Brain Behav. 2022. PMID: 34978387 Free PMC article.
-
Limb-girdle myasthenia with tubular aggregates associated with novel GFPT1 mutations.Muscle Nerve. 2012 Oct;46(4):600-4. doi: 10.1002/mus.23451. Muscle Nerve. 2012. PMID: 22987706 Review.
-
Congenital myasthenic syndromes.Handb Clin Neurol. 2013;113:1469-80. doi: 10.1016/B978-0-444-59565-2.00016-2. Handb Clin Neurol. 2013. PMID: 23622369 Review.
Cited by
-
Novel Genetic and Biochemical Insights into the Spectrum of NEFL-Associated Phenotypes.J Neuromuscul Dis. 2024;11(3):625-645. doi: 10.3233/JND-230230. J Neuromuscul Dis. 2024. PMID: 38578900 Free PMC article. Review.
-
STIM1/ORAI1 Loss-of-Function and Gain-of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases.Front Physiol. 2020 Nov 4;11:604941. doi: 10.3389/fphys.2020.604941. eCollection 2020. Front Physiol. 2020. PMID: 33250786 Free PMC article. Review.
-
GFPT1-related congenital myasthenic syndrome misdiagnosed as myopathy: clinical and genetic insights.Acta Neurol Belg. 2025 Apr 15. doi: 10.1007/s13760-025-02783-2. Online ahead of print. Acta Neurol Belg. 2025. PMID: 40232331
-
The Hexosamine Biosynthesis Pathway: Regulation and Function.Genes (Basel). 2023 Apr 18;14(4):933. doi: 10.3390/genes14040933. Genes (Basel). 2023. PMID: 37107691 Free PMC article. Review.
-
A cohort of GFPT1 related congenital myasthenic syndrome in China: high frequency of c.331 c > t variant.Orphanet J Rare Dis. 2025 May 29;20(1):259. doi: 10.1186/s13023-025-03823-z. Orphanet J Rare Dis. 2025. PMID: 40442802 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
