

Pathogenic variant in EPHB4 results in central conducting lymphatic anomaly
- PMID: 29905864
- PMCID: PMC7190898
- DOI: 10.1093/hmg/ddy218
Pathogenic variant in EPHB4 results in central conducting lymphatic anomaly
Abstract
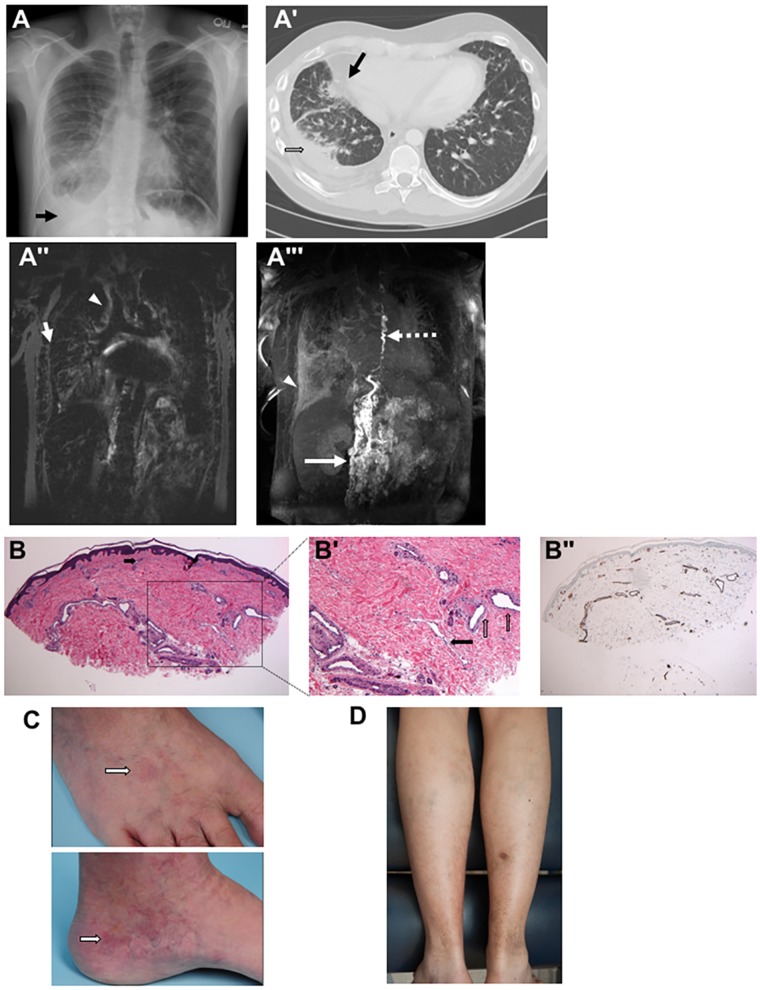
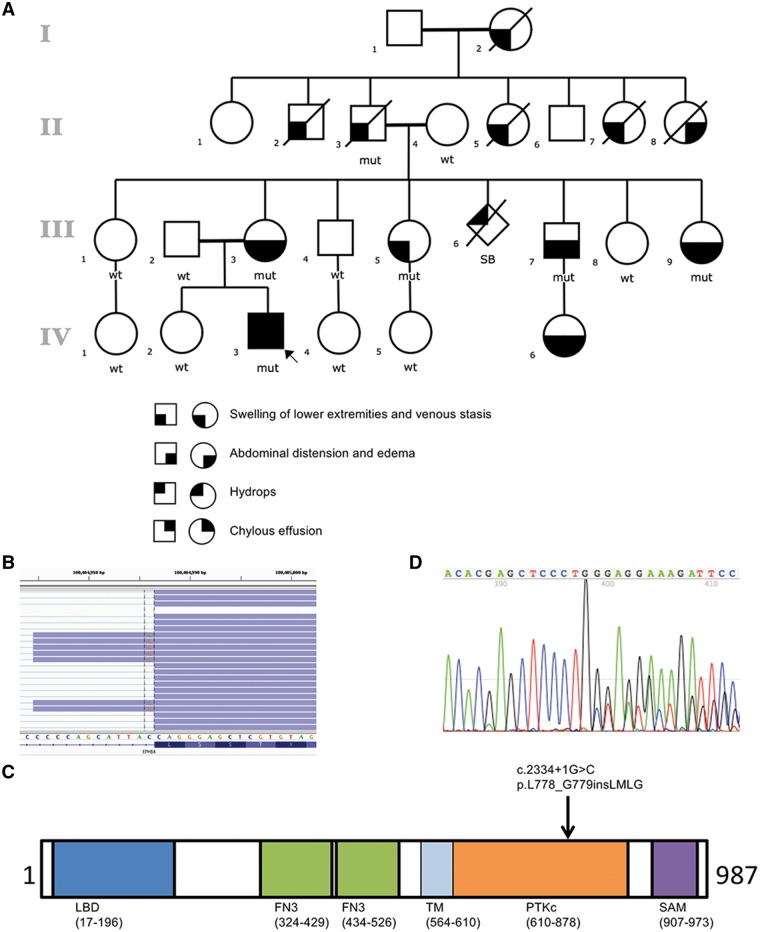
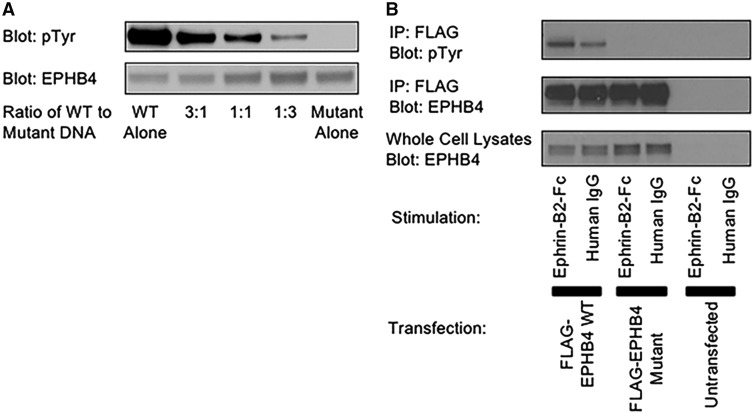
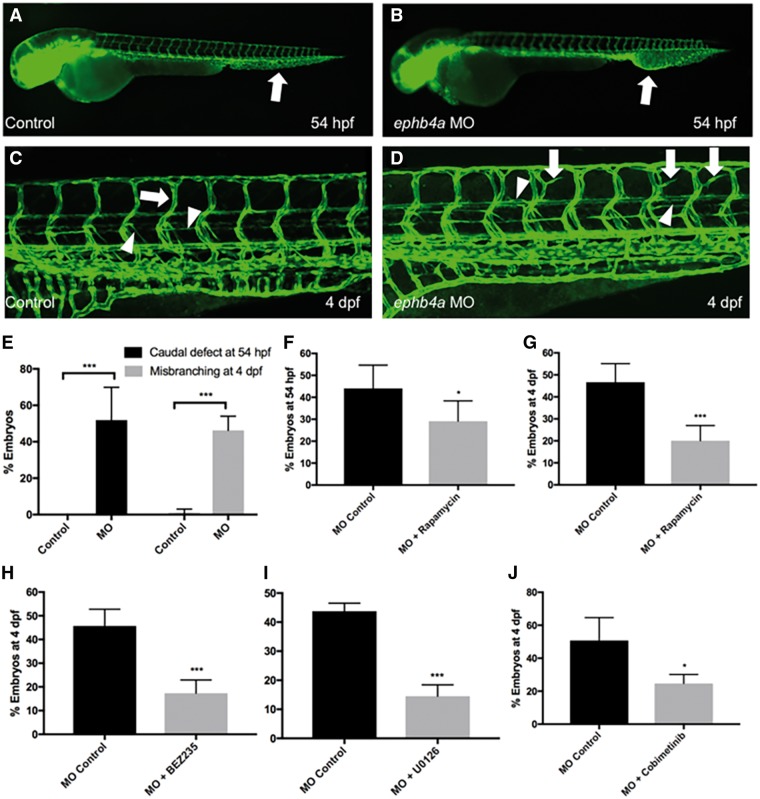
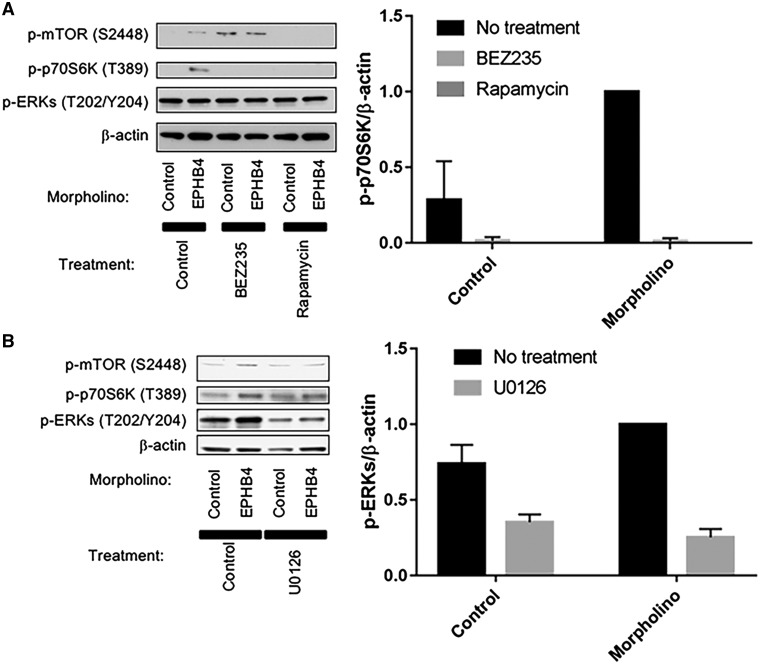
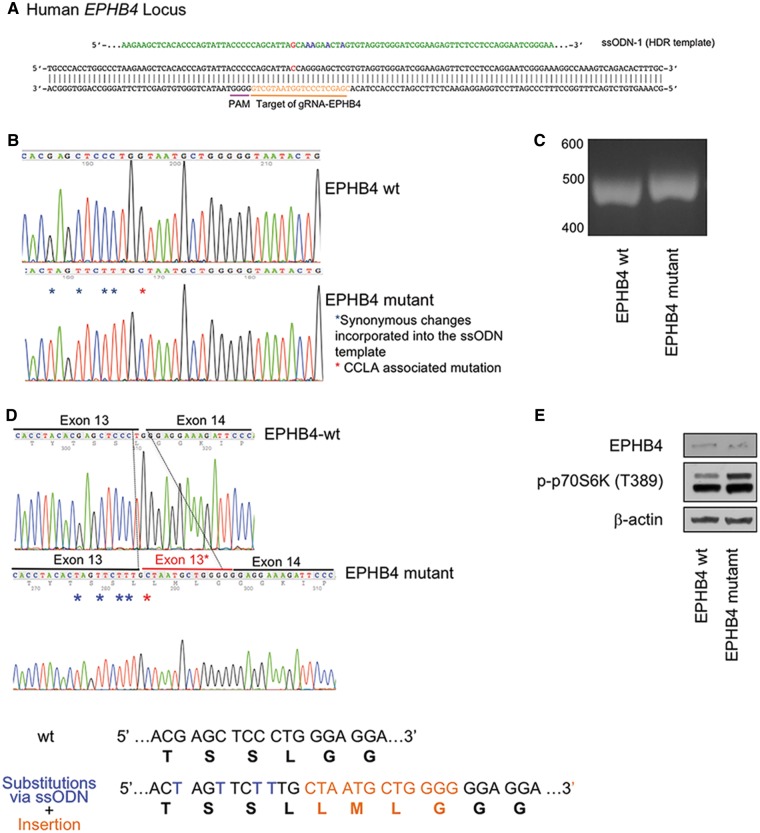
Central conducting lymphatic anomaly (CCLA) is one of the complex lymphatic anomalies characterized by dilated lymphatic channels, lymphatic channel dysmotility and distal obstruction affecting lymphatic drainage. We performed whole exome sequencing (WES) of DNA from a four-generation pedigree and examined the consequences of the variant by transfection of mammalian cells and morpholino and rescue studies in zebrafish. WES revealed a heterozygous mutation in EPHB4 (RefSeq NM_004444.4; c.2334 + 1G>C) and RNA-Seq demonstrated that the EPHB4 mutation destroys the normal donor site, which leads to the use of a cryptic splice donor that results in retention of the intervening 12-bp intron sequence. Transient co-expression of the wild-type and mutant EPHB4 proteins showed reduced phosphorylation of tyrosine, consistent with a loss-of-function effect. Zebrafish ephb4a morpholino resulted in vessel misbranching and deformities in the lymphatic vessel development, indicative of possible differentiation defects in lymphatic vessels, mimicking the lymphatic presentations of the patients. Immunoblot analysis using zebrafish lysates demonstrated over-activation of mTORC1 as a consequence of reduced EPHB4 signaling. Strikingly, drugs that inhibit mTOR signaling or RAS-MAPK signaling effectively rescued the misbranching phenotype in a comparable manner. Moreover, knock-in of EPHB4 mutation in HEK293T cells also induced mTORC1 activity. Our data demonstrate the pathogenicity of the identified EPHB4 mutation as a novel cause of CCLA and suggesting that ERK inhibitors may have therapeutic benefits in such patients with complex lymphatic anomalies.
Figures
References
-
- Alitalo K., Tammela T., Petrova T.V. (2005) Lymphangiogenesis in development and human disease. Nature, 438, 946–953. - PubMed
-
- Adams R.H., Alitalo K. (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell. Biol., 8, 464–478. - PubMed
-
- Tammela T., Alitalo K. (2010) Lymphangiogenesis: molecular mechanisms and future promise. Cell, 140, 460–476. - PubMed
-
- Alitalo K. (2011) The lymphatic vasculature in disease. Nat. Med., 17, 1371–1380. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
