

Oxidation Resistance 1 Modulates Glycolytic Pathways in the Cerebellum via an Interaction with Glucose-6-Phosphate Isomerase
- PMID: 29905912
- PMCID: PMC6368252
- DOI: 10.1007/s12035-018-1174-x
Oxidation Resistance 1 Modulates Glycolytic Pathways in the Cerebellum via an Interaction with Glucose-6-Phosphate Isomerase
Abstract
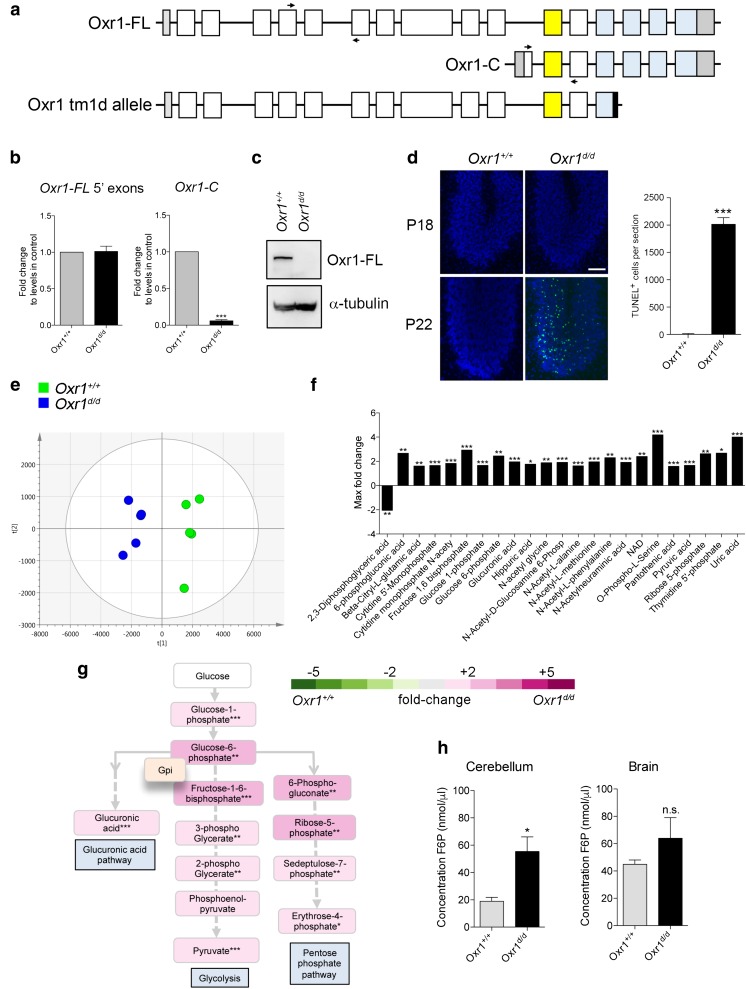
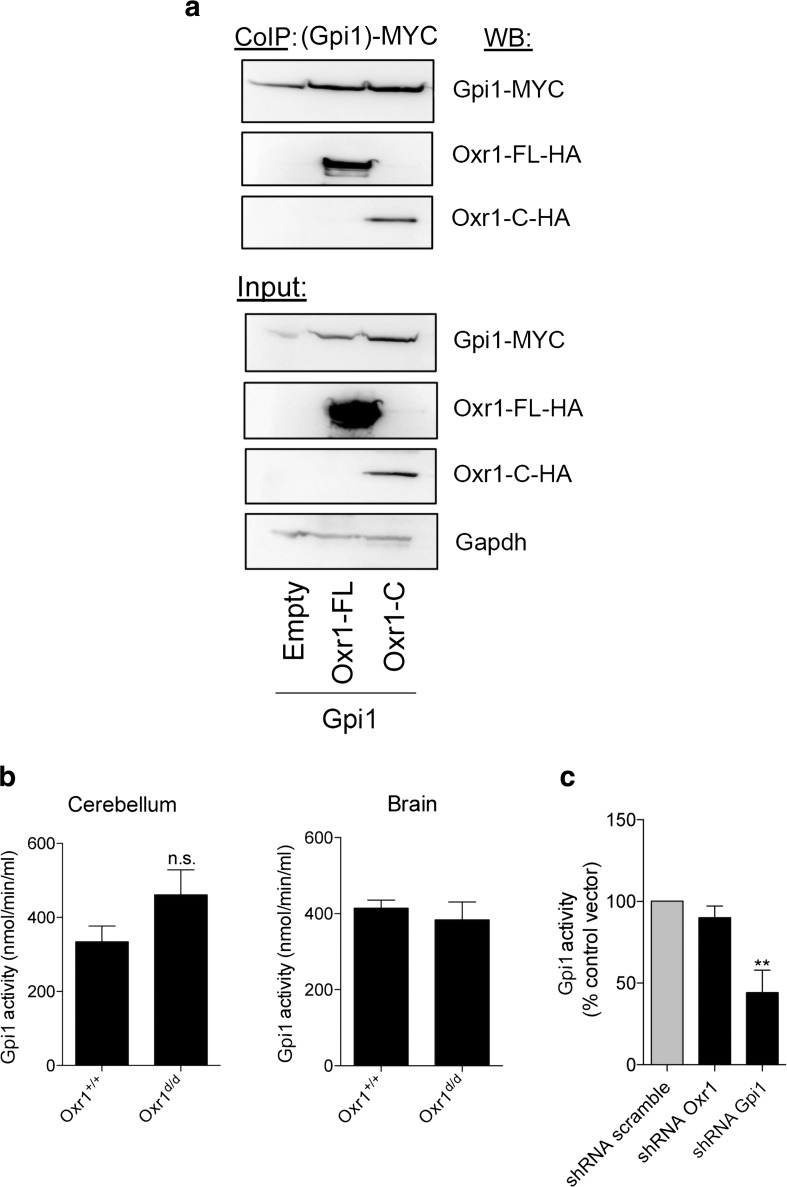
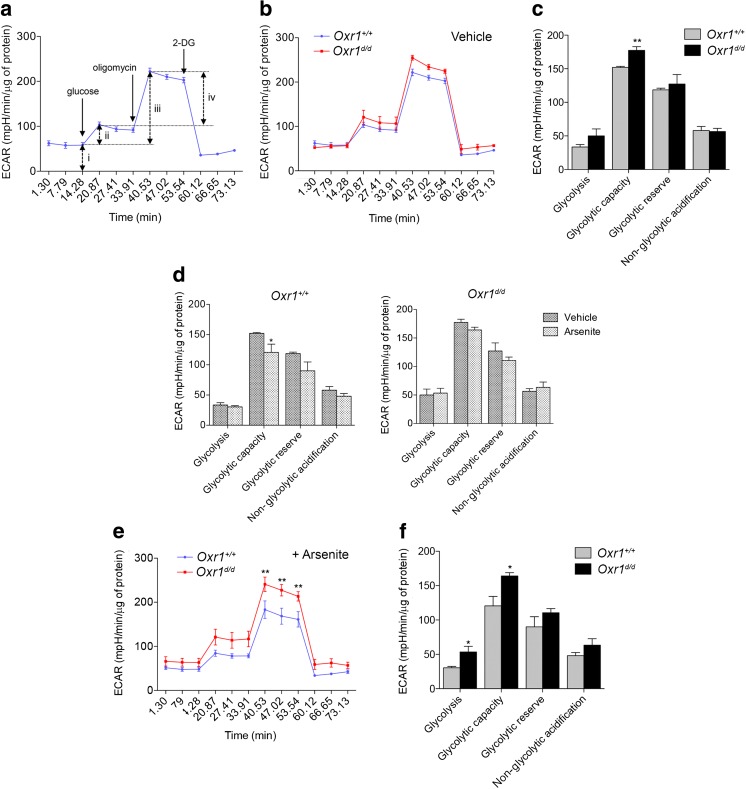
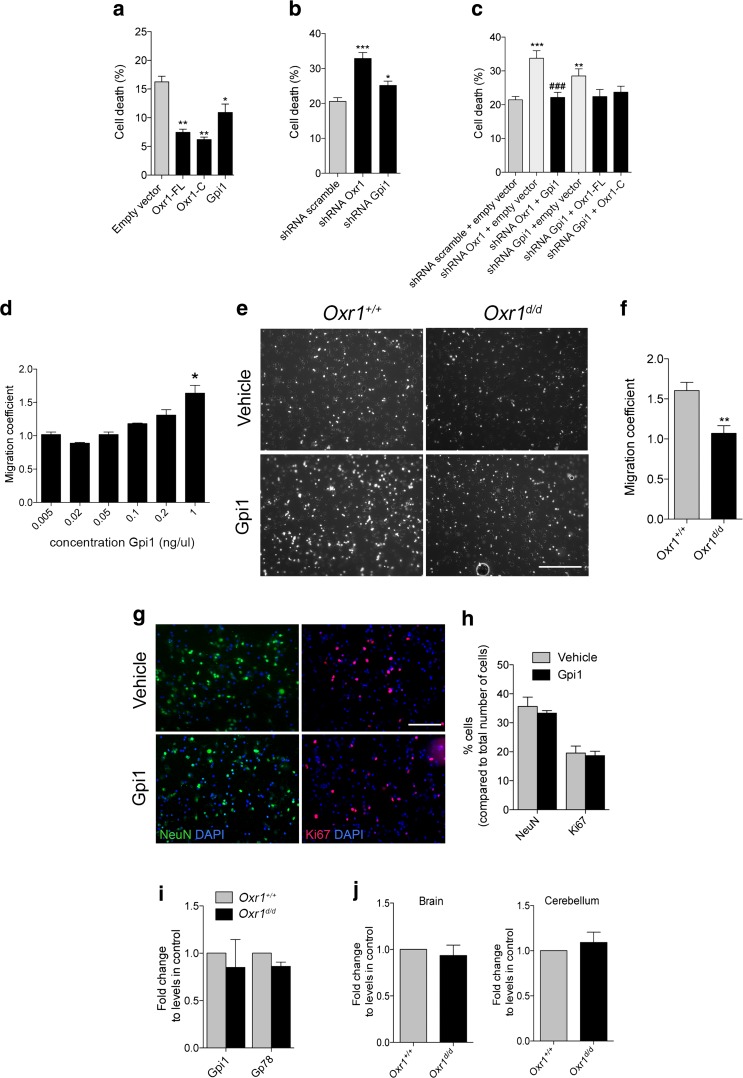
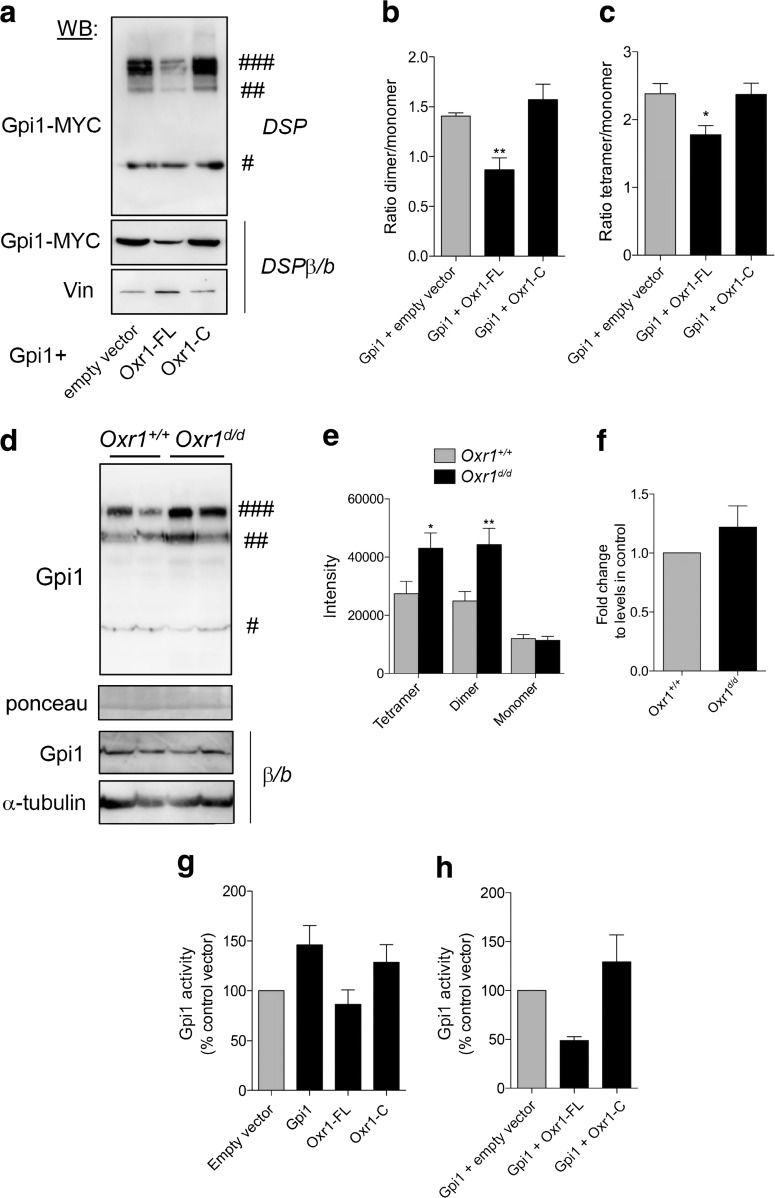
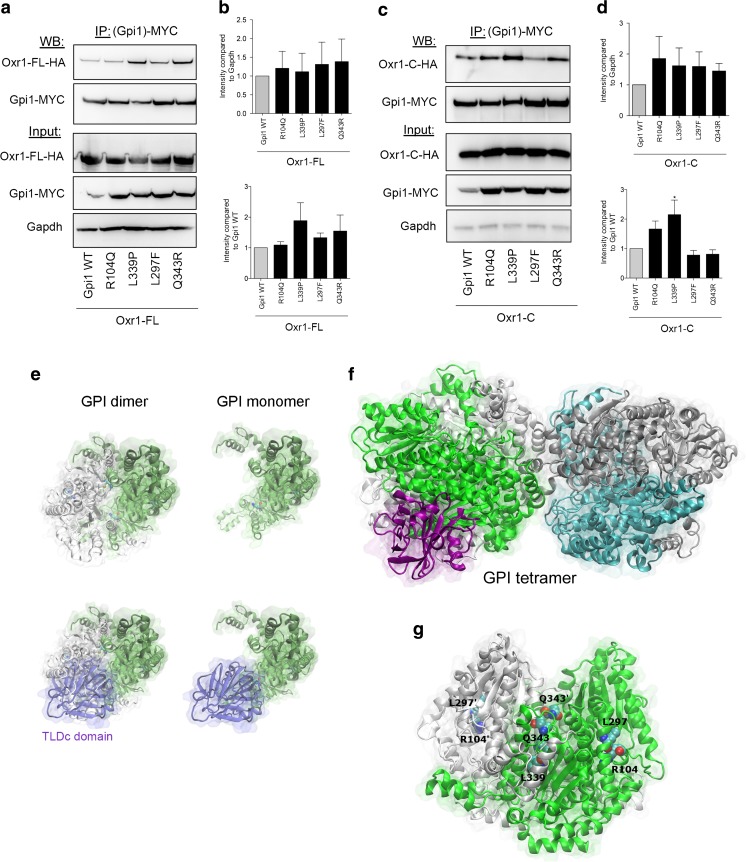
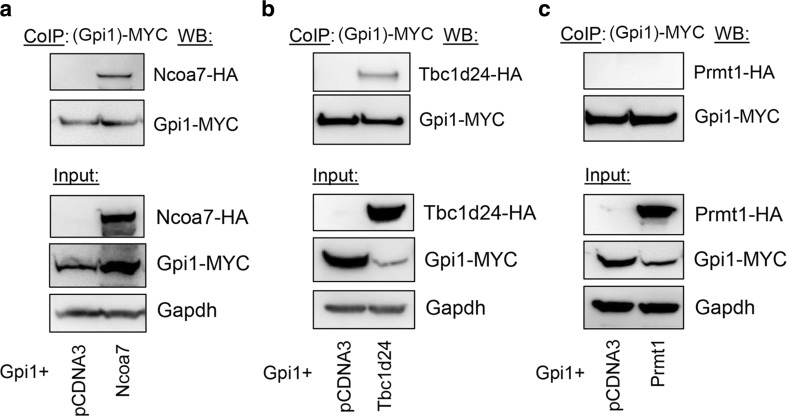
Glucose metabolism is essential for the brain: it not only provides the required energy for cellular function and communication but also participates in balancing the levels of oxidative stress in neurons. Defects in glucose metabolism have been described in neurodegenerative disease; however, it remains unclear how this fundamental process contributes to neuronal cell death in these disorders. Here, we investigated the molecular mechanisms driving the selective neurodegeneration in an ataxic mouse model lacking oxidation resistance 1 (Oxr1) and discovered an unexpected function for this protein as a regulator of the glycolytic enzyme, glucose-6-phosphate isomerase (GPI/Gpi1). Initially, we present a dysregulation of metabolites of glucose metabolism at the pre-symptomatic stage in the Oxr1 knockout cerebellum. We then demonstrate that Oxr1 and Gpi1 physically and functionally interact and that the level of Gpi1 oligomerisation is disrupted when Oxr1 is deleted in vivo. Furthermore, we show that Oxr1 modulates the additional and less well-understood roles of Gpi1 as a cytokine and neuroprotective factor. Overall, our data identify a new molecular function for Oxr1, establishing this protein as important player in neuronal survival, regulating both oxidative stress and glucose metabolism in the brain.
Keywords: Cerebellum; Glucose metabolism; Mouse; Neurodegeneration; Oxidative stress.
Figures
Similar articles
-
Preventing Neurodegeneration by Controlling Oxidative Stress: The Role of OXR1.Front Neurosci. 2020 Dec 15;14:611904. doi: 10.3389/fnins.2020.611904. eCollection 2020. Front Neurosci. 2020. PMID: 33384581 Free PMC article. Review.
-
Oxidation resistance 1 regulates post-translational modifications of peroxiredoxin 2 in the cerebellum.Free Radic Biol Med. 2019 Jan;130:151-162. doi: 10.1016/j.freeradbiomed.2018.10.447. Epub 2018 Oct 31. Free Radic Biol Med. 2019. PMID: 30389497 Free PMC article.
-
The antioxidant protein Oxr1 influences aspects of mitochondrial morphology.Free Radic Biol Med. 2016 Jun;95:255-67. doi: 10.1016/j.freeradbiomed.2016.03.029. Epub 2016 Mar 29. Free Radic Biol Med. 2016. PMID: 27036366 Free PMC article.
-
Oxr1 is essential for protection against oxidative stress-induced neurodegeneration.PLoS Genet. 2011 Oct;7(10):e1002338. doi: 10.1371/journal.pgen.1002338. Epub 2011 Oct 20. PLoS Genet. 2011. PMID: 22028674 Free PMC article.
-
Glucose, glycolysis, and neurodegenerative diseases.J Cell Physiol. 2020 Nov;235(11):7653-7662. doi: 10.1002/jcp.29682. Epub 2020 Apr 2. J Cell Physiol. 2020. PMID: 32239718 Review.
Cited by
-
Preventing Neurodegeneration by Controlling Oxidative Stress: The Role of OXR1.Front Neurosci. 2020 Dec 15;14:611904. doi: 10.3389/fnins.2020.611904. eCollection 2020. Front Neurosci. 2020. PMID: 33384581 Free PMC article. Review.
-
OXR2 Increases Plant Defense against a Hemibiotrophic Pathogen via the Salicylic Acid Pathway.Plant Physiol. 2020 Oct;184(2):1112-1127. doi: 10.1104/pp.19.01351. Epub 2020 Jul 29. Plant Physiol. 2020. PMID: 32727912 Free PMC article.
-
The role of glycolytic metabolic pathways in cardiovascular disease and potential therapeutic approaches.Basic Res Cardiol. 2023 Nov 8;118(1):48. doi: 10.1007/s00395-023-01018-w. Basic Res Cardiol. 2023. PMID: 37938421 Free PMC article. Review.
-
Bioinformatics analysis of differentially expressed genes and identification of an miRNA-mRNA network associated with entorhinal cortex and hippocampus in Alzheimer's disease.Hereditas. 2021 Jul 9;158(1):25. doi: 10.1186/s41065-021-00190-0. Hereditas. 2021. PMID: 34243818 Free PMC article.
-
Neuronal over-expression of Oxr1 is protective against ALS-associated mutant TDP-43 mislocalisation in motor neurons and neuromuscular defects in vivo.Hum Mol Genet. 2019 Nov 1;28(21):3584-3599. doi: 10.1093/hmg/ddz190. Hum Mol Genet. 2019. PMID: 31642482 Free PMC article.
References
-
- Clarke DD, Sokoloff L (1999) Chapter 31. Circulation and energy metabolism of the brain. In: Lippincott P (ed) Basic neurochemistry: molecular, cellular and medical aspects. pp 637–670
-
- Cistaro A, Valentini MC, Chiò A, Nobili F, Calvo A, Moglia C, Montuschi A, Morbelli S, Salmaso D, Fania P, Carrara G, Pagani M. Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur J Nucl Med Mol Imaging. 2012;39(2):251–259. doi: 10.1007/s00259-011-1979-6. - DOI - PubMed
-
- López-Mora DA, Camacho V, Pérez-Pérez J, Martínez-Horta S, Fernández A, Sampedro F, Montes A, Lozano-Martínez GA, Gómez-Anson B, Kulisevsky J, Carrió I. Striatal hypometabolism in premanifest and manifest Huntington’s disease patients. Eur J Nucl Med Mol Imaging. 2016;43(12):2183–2189. doi: 10.1007/s00259-016-3445-y. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
