

Tellurium notebooks-An environment for reproducible dynamical modeling in systems biology
- PMID: 29906293
- PMCID: PMC6021116
- DOI: 10.1371/journal.pcbi.1006220
Tellurium notebooks-An environment for reproducible dynamical modeling in systems biology
Abstract
The considerable difficulty encountered in reproducing the results of published dynamical models limits validation, exploration and reuse of this increasingly large biomedical research resource. To address this problem, we have developed Tellurium Notebook, a software system for model authoring, simulation, and teaching that facilitates building reproducible dynamical models and reusing models by 1) providing a notebook environment which allows models, Python code, and narrative to be intermixed, 2) supporting the COMBINE archive format during model development for capturing model information in an exchangeable format and 3) enabling users to easily simulate and edit public COMBINE-compliant models from public repositories to facilitate studying model dynamics, variants and test cases. Tellurium Notebook, a Python-based Jupyter-like environment, is designed to seamlessly inter-operate with these community standards by automating conversion between COMBINE standards formulations and corresponding in-line, human-readable representations. Thus, Tellurium brings to systems biology the strategy used by other literate notebook systems such as Mathematica. These capabilities allow users to edit every aspect of the standards-compliant models and simulations, run the simulations in-line, and re-export to standard formats. We provide several use cases illustrating the advantages of our approach and how it allows development and reuse of models without requiring technical knowledge of standards. Adoption of Tellurium should accelerate model development, reproducibility and reuse.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
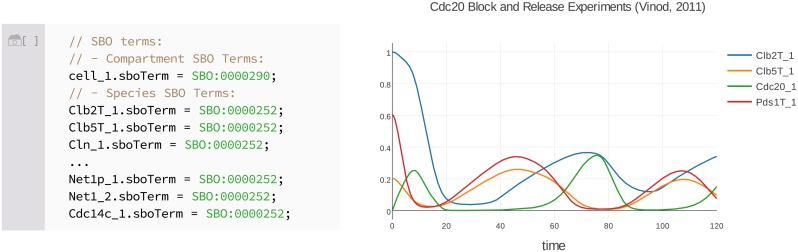
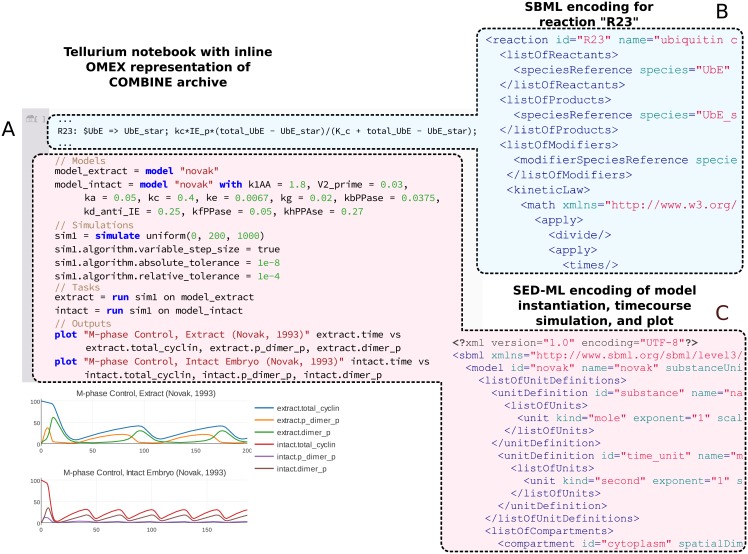
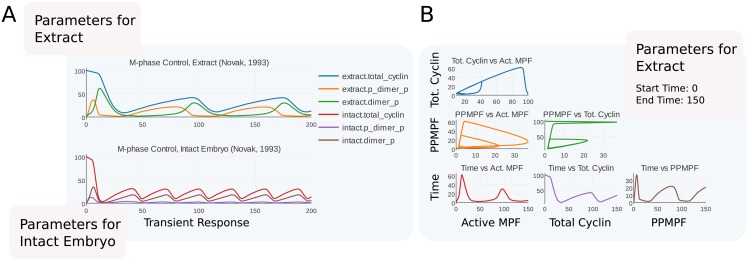
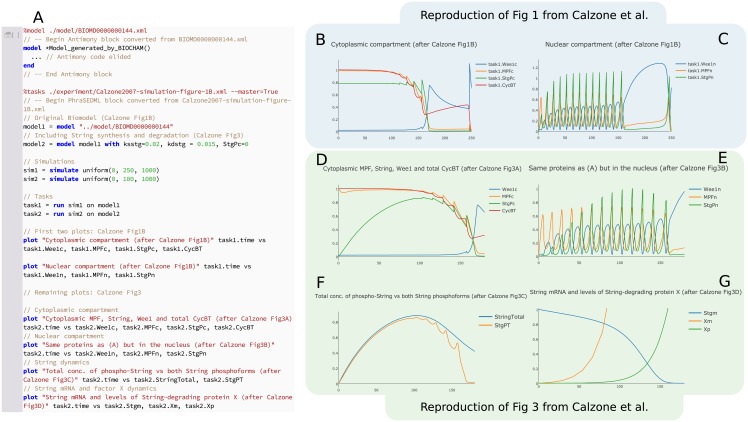
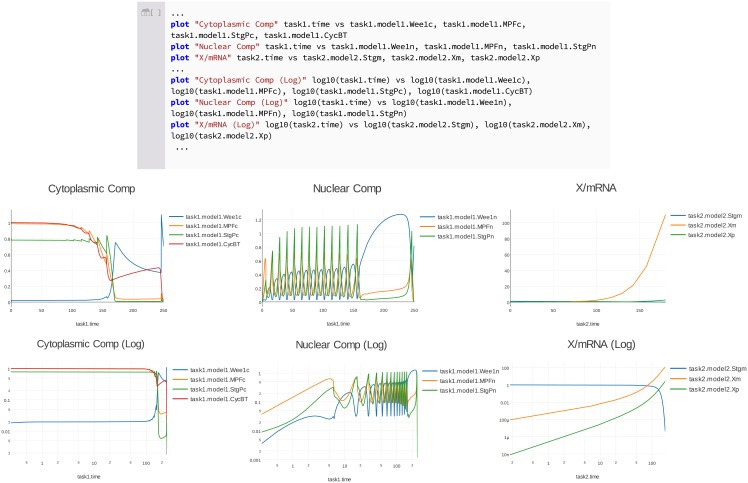
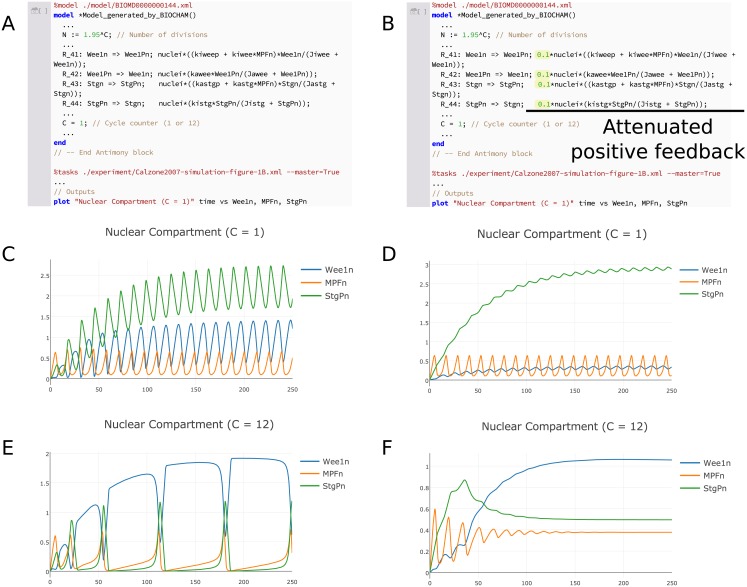
References
-
- Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B, et al. A whole-cell computational model predicts phenotype from genotype. Cell. 2012;150(2):389–401. doi: 10.1016/j.cell.2012.05.044 - DOI - PMC - PubMed
-
- Millard P, Smallbone K, Mendes P. Metabolic regulation is sufficient for global and robust coordination of glucose uptake, catabolism, energy production and growth in Escherichia coli. PLoS computational biology. 2017;13(2):e1005396 doi: 10.1371/journal.pcbi.1005396 - DOI - PMC - PubMed
-
- Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nature reviews Drug discovery. 2011;10(9):712–712. doi: 10.1038/nrd3439-c1 - DOI - PubMed
-
- Mobley A, Linder SK, Braeuer R, Ellis LM, Zwelling L. A survey on data reproducibility in cancer research provides insights into our limited ability to translate findings from the laboratory to the clinic. PLoS One. 2013;8(5):e63221 doi: 10.1371/journal.pone.0063221 - DOI - PMC - PubMed
-
- Peng GC. Moving Toward Model Reproducibility and Reusability. IEEE Transactions on Biomedical Engineering. 2016;63(10):1997–1998. doi: 10.1109/TBME.2016.2603418 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
