

Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer
- PMID: 29906450
- PMCID: PMC6084431
- DOI: 10.1016/j.cell.2018.04.034
Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer
Abstract
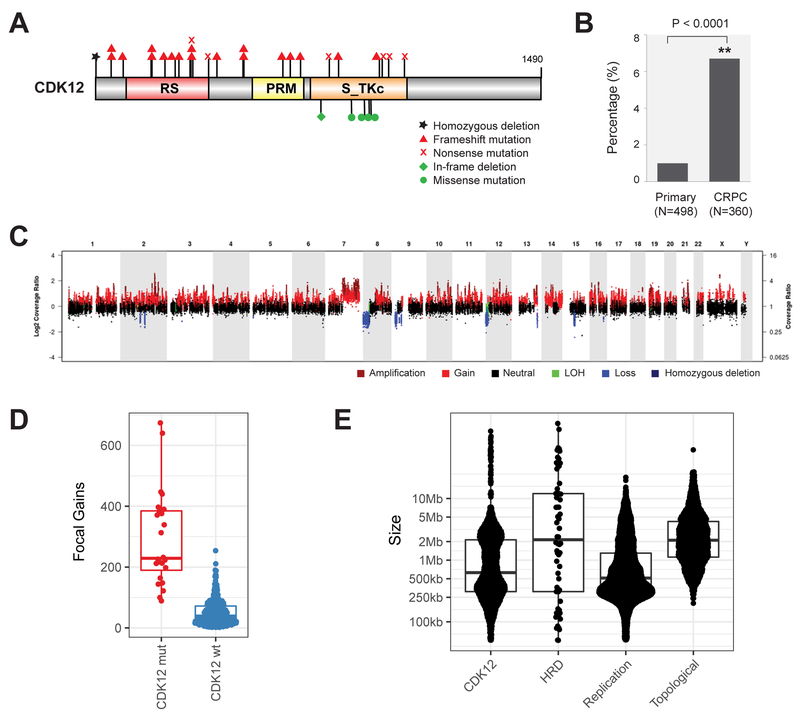
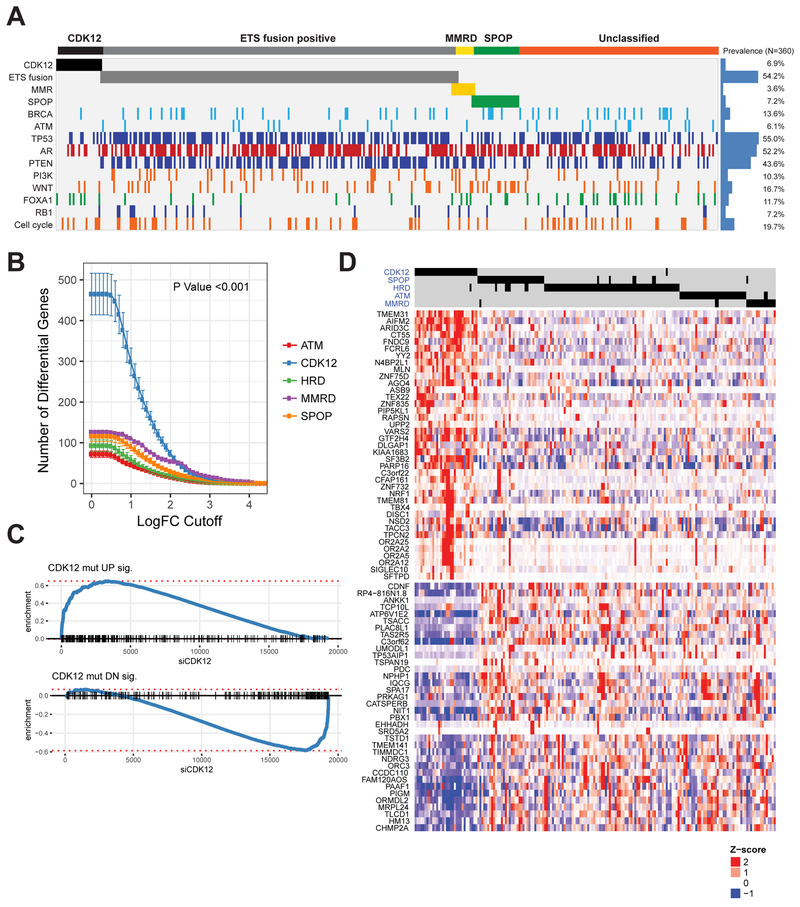
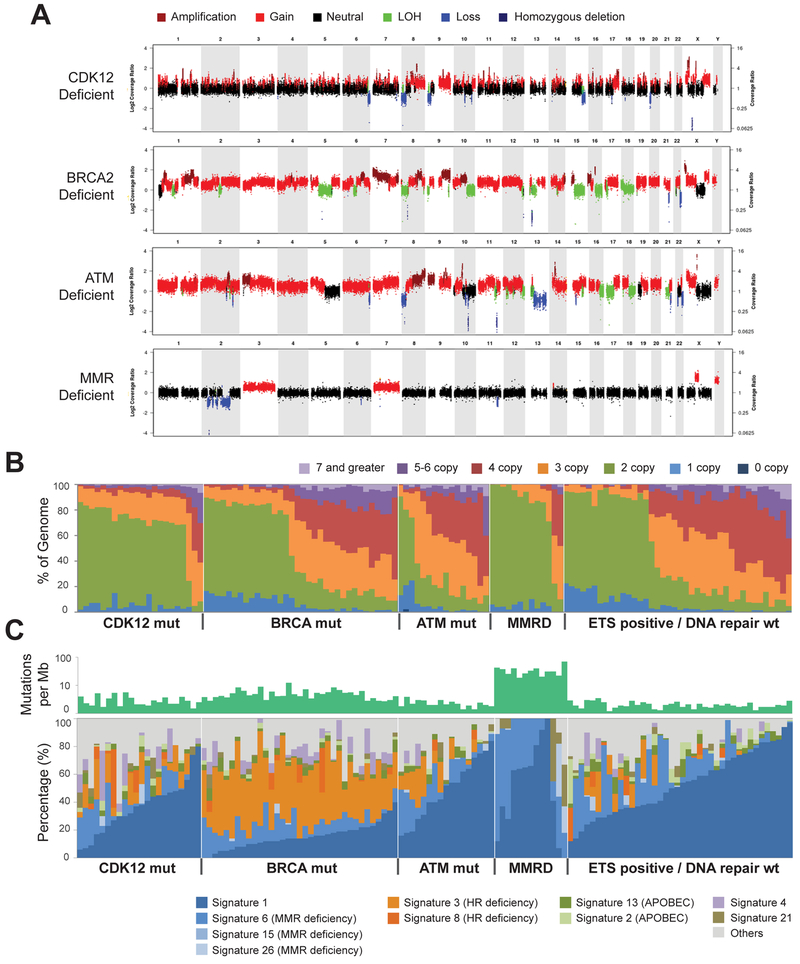
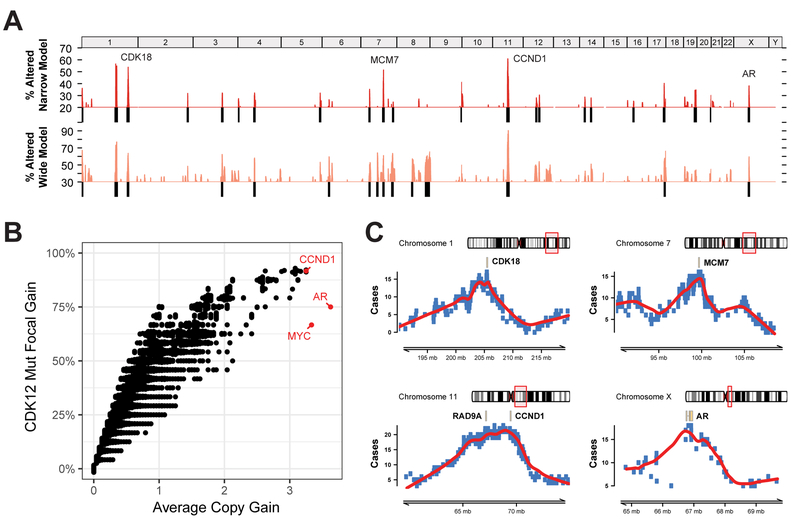
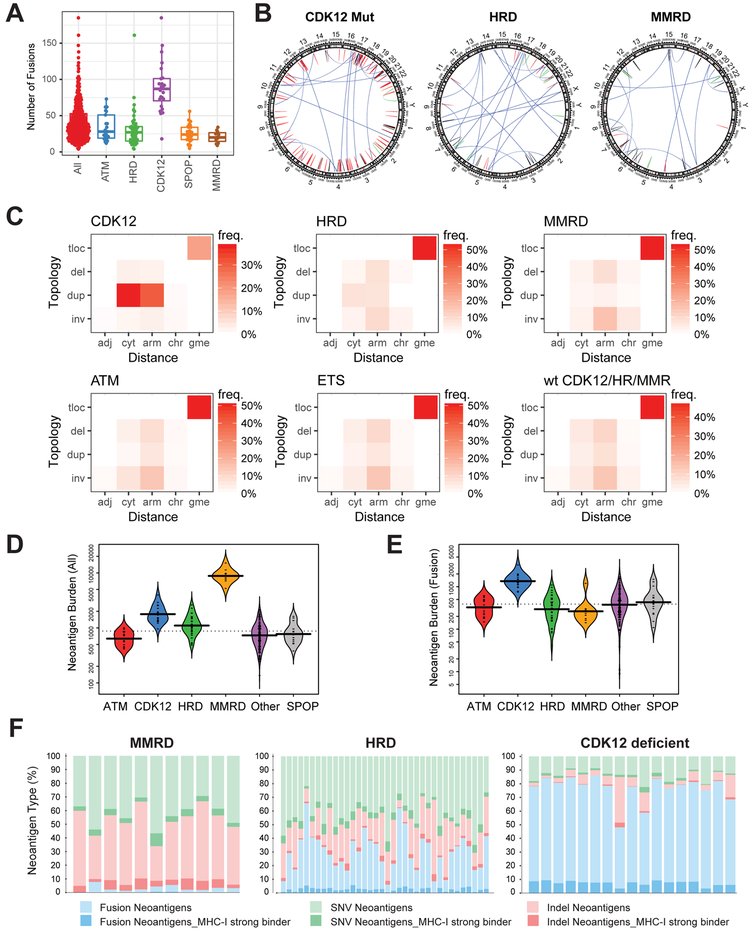
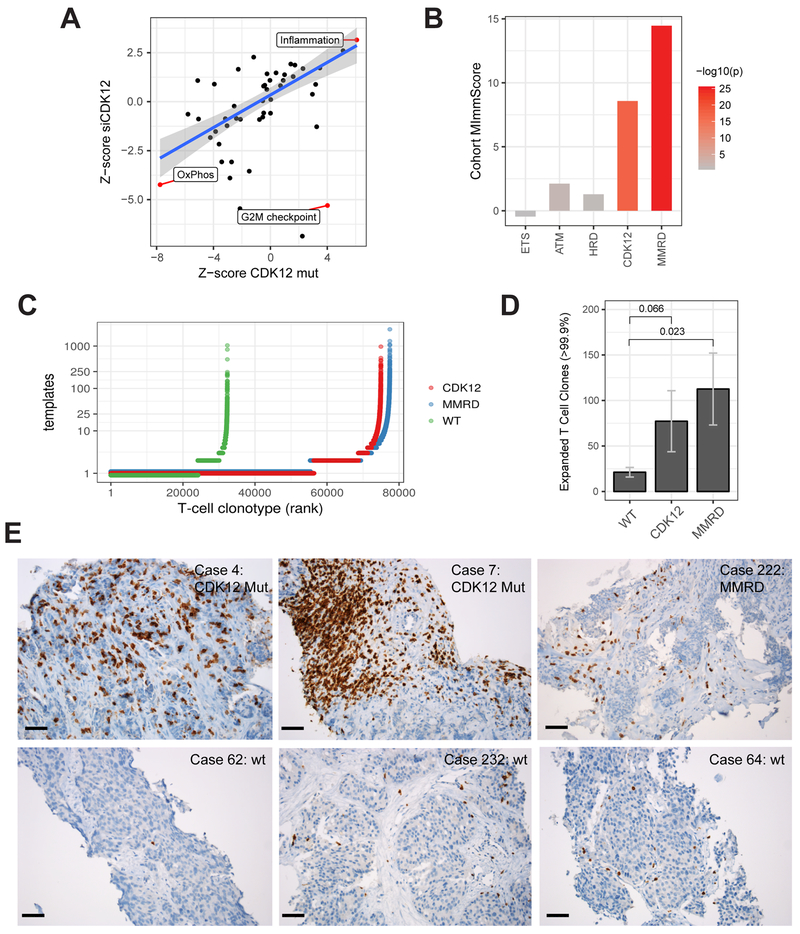
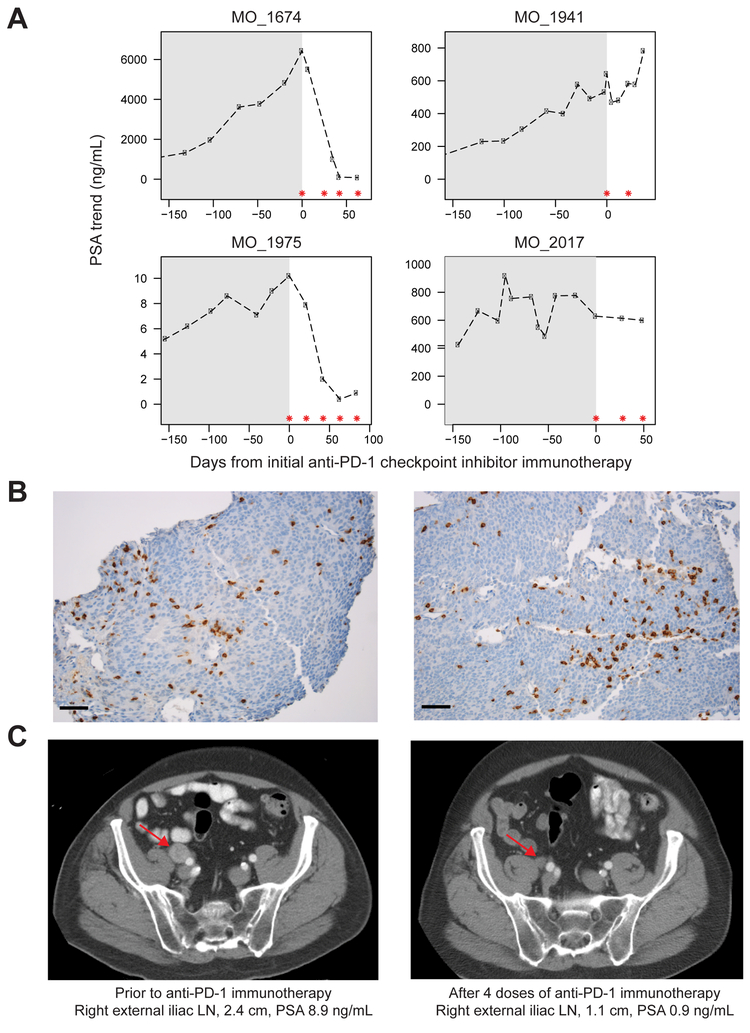
Using integrative genomic analysis of 360 metastatic castration-resistant prostate cancer (mCRPC) samples, we identified a novel subtype of prostate cancer typified by biallelic loss of CDK12 that is mutually exclusive with tumors driven by DNA repair deficiency, ETS fusions, and SPOP mutations. CDK12 loss is enriched in mCRPC relative to clinically localized disease and characterized by focal tandem duplications (FTDs) that lead to increased gene fusions and marked differential gene expression. FTDs associated with CDK12 loss result in highly recurrent gains at loci of genes involved in the cell cycle and DNA replication. CDK12 mutant cases are baseline diploid and do not exhibit DNA mutational signatures linked to defects in homologous recombination. CDK12 mutant cases are associated with elevated neoantigen burden ensuing from fusion-induced chimeric open reading frames and increased tumor T cell infiltration/clonal expansion. CDK12 inactivation thereby defines a distinct class of mCRPC that may benefit from immune checkpoint immunotherapy.
Keywords: CDK12; focal tandem duplications; gene fusions; immunotherapy; metastatic castration-resistant prostate cancer; neoantigens.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
Comment in
-
New genomic drivers identified.Nat Rev Urol. 2018 Sep;15(9):525. doi: 10.1038/s41585-018-0057-2. Nat Rev Urol. 2018. PMID: 29977054 No abstract available.
-
CDK12 Changes Telling in Prostate Cancer.Cancer Discov. 2018 Sep;8(9):1055. doi: 10.1158/2159-8290.CD-NB2018-093. Epub 2018 Jul 13. Cancer Discov. 2018. PMID: 30006379
References
-
- Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, Danila D, Rathkopf D, Morris M, Slovin S, et al. (2017). Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol 2017. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
