

A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs
- PMID: 29907613
- PMCID: PMC6071642
- DOI: 10.1101/gr.233049.117
A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs
Abstract
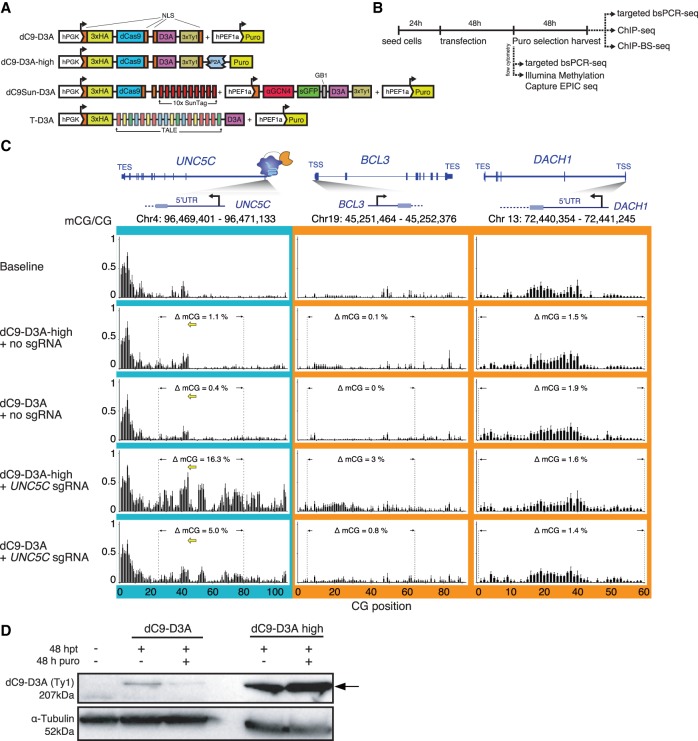
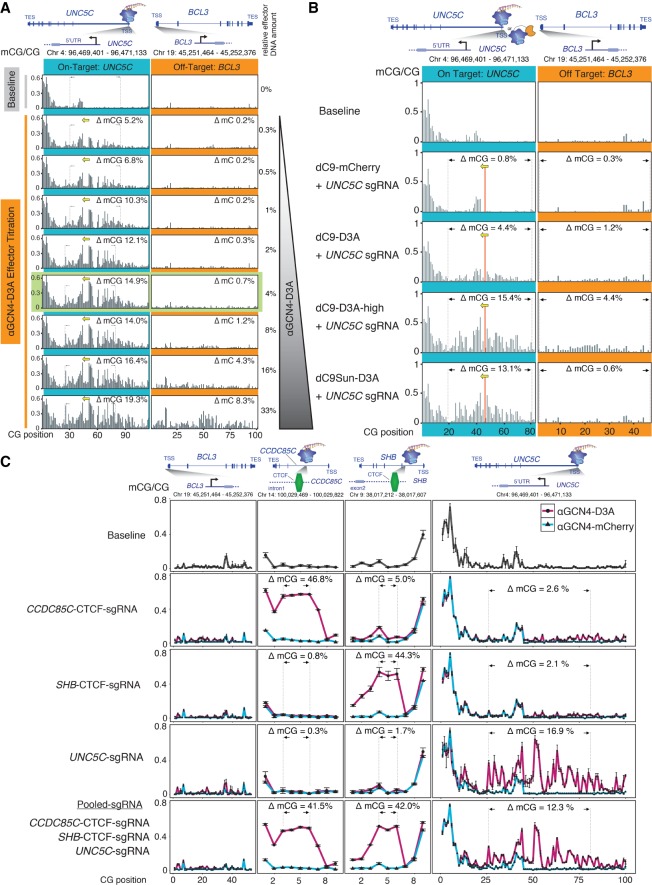
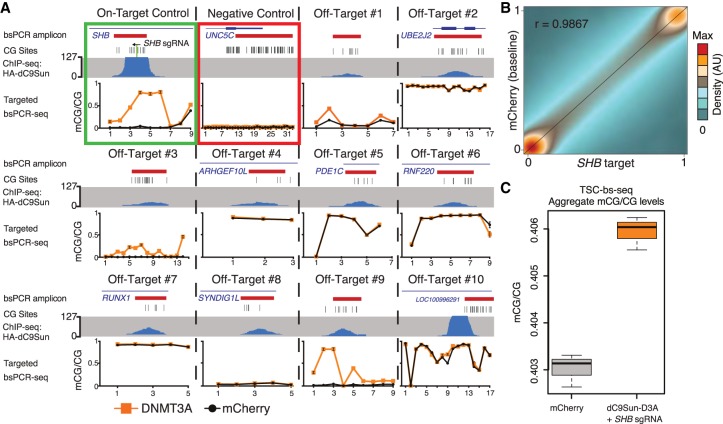
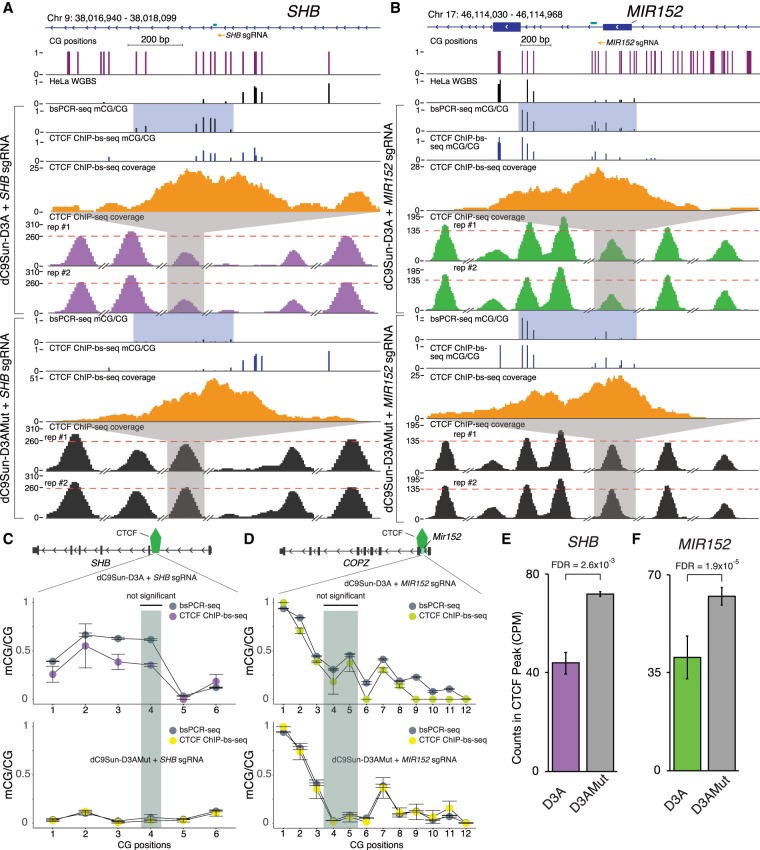
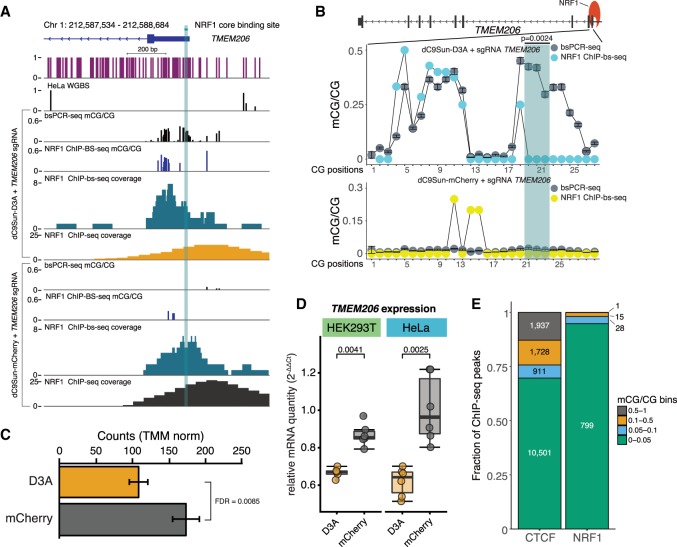
Detection of DNA methylation in the genome has been possible for decades; however, the ability to deliberately and specifically manipulate local DNA methylation states in the genome has been extremely limited. Consequently, this has impeded our understanding of the direct effect of DNA methylation on transcriptional regulation and transcription factor binding in the native chromatin context. Thus, highly specific targeted epigenome editing tools are needed to address this. Recent adaptations of genome editing technologies, including fusion of the DNMT3A DNA methyltransferase catalytic domain to catalytically inactive Cas9 (dC9-D3A), have aimed to alter DNA methylation at desired loci. Here, we show that these tools exhibit consistent off-target DNA methylation deposition in the genome, limiting their capabilities to unambiguously assess the functional consequences of DNA methylation. To address this, we developed a modular dCas9-SunTag (dC9Sun-D3A) system that can recruit multiple DNMT3A catalytic domains to a target site for editing DNA methylation. dC9Sun-D3A is tunable, specific, and exhibits much higher induction of DNA methylation at target sites than the dC9-D3A direct fusion protein. Importantly, genome-wide characterization of dC9Sun-D3A binding sites and DNA methylation revealed minimal off-target protein binding and induction of DNA methylation with dC9Sun-D3A, compared to pervasive off-target methylation by dC9-D3A. Furthermore, we used dC9Sun-D3A to demonstrate the binding sensitivity to DNA methylation for CTCF and NRF1 in situ. Overall, this modular dC9Sun-D3A system enables precise DNA methylation deposition with the lowest off-target DNA methylation levels reported to date, allowing accurate functional determination of the role of DNA methylation at single loci.
© 2018 Pflueger et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Bernstein BE, Meissner A, Lander ES. 2007. The mammalian epigenome. Cell 128: 669–681. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials