

Determinants of Base-Pair Substitution Patterns Revealed by Whole-Genome Sequencing of DNA Mismatch Repair Defective Escherichia coli
- PMID: 29907647
- PMCID: PMC6063221
- DOI: 10.1534/genetics.118.301237
Determinants of Base-Pair Substitution Patterns Revealed by Whole-Genome Sequencing of DNA Mismatch Repair Defective Escherichia coli
Abstract
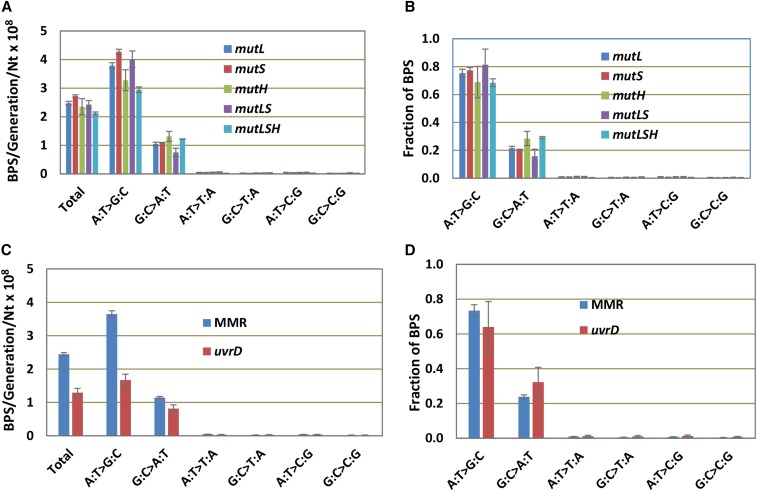
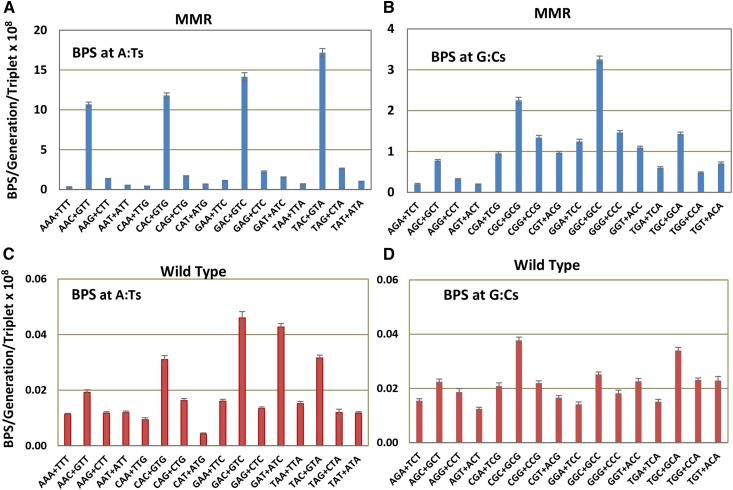
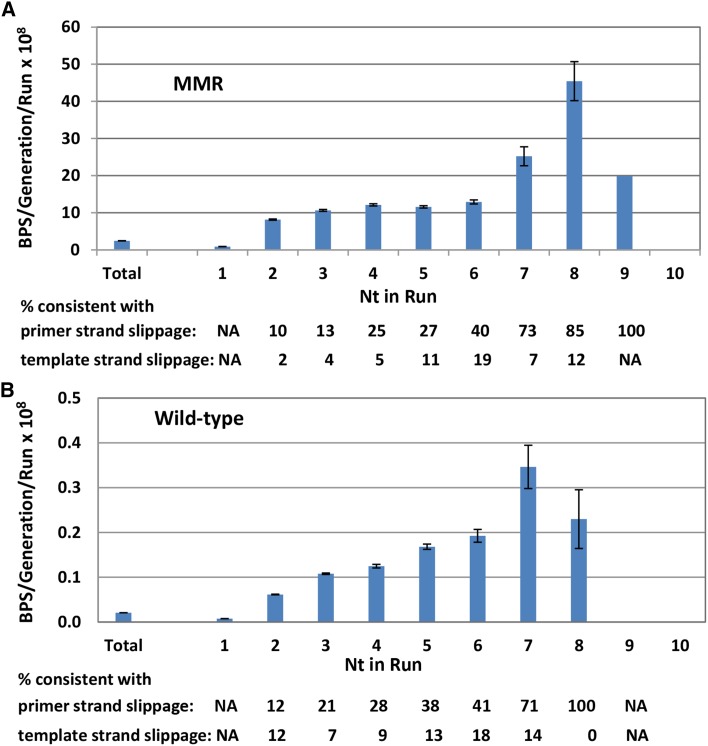
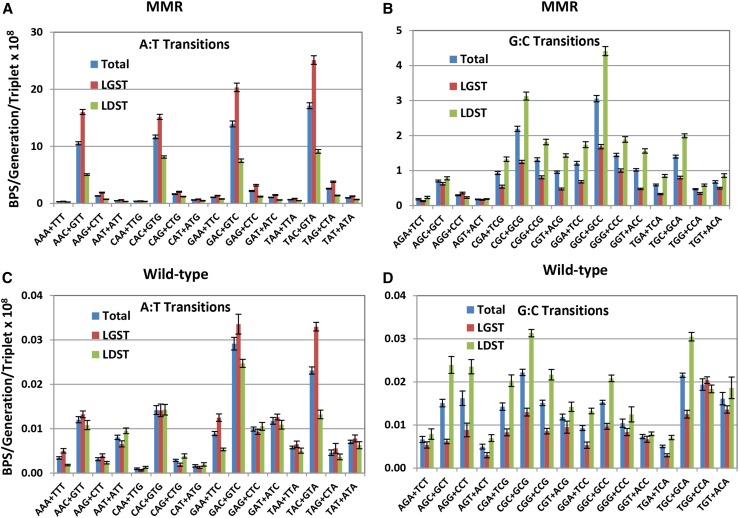
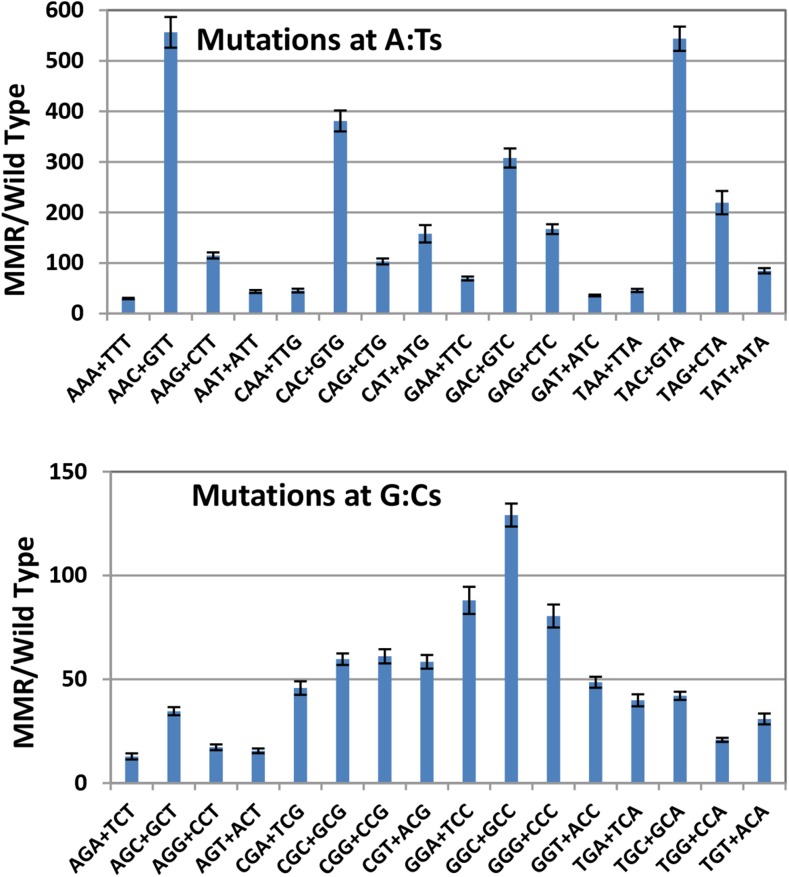
Mismatch repair (MMR) is a major contributor to replication fidelity, but its impact varies with sequence context and the nature of the mismatch. Mutation accumulation experiments followed by whole-genome sequencing of MMR-defective Escherichia coli strains yielded ≈30,000 base-pair substitutions (BPSs), revealing mutational patterns across the entire chromosome. The BPS spectrum was dominated by A:T to G:C transitions, which occurred predominantly at the center base of 5'NAC3'+5'GTN3' triplets. Surprisingly, growth on minimal medium or at low temperature attenuated these mutations. Mononucleotide runs were also hotspots for BPSs, and the rate at which these occurred increased with run length. Comparison with ≈2000 BPSs accumulated in MMR-proficient strains revealed that both kinds of hotspots appeared in the wild-type spectrum and so are likely to be sites of frequent replication errors. In MMR-defective strains transitions were strand biased, occurring twice as often when A and C rather than T and G were on the lagging-strand template. Loss of nucleotide diphosphate kinase increases the cellular concentration of dCTP, which resulted in increased rates of mutations due to misinsertion of C opposite A and T. In an mmr ndk double mutant strain, these mutations were more frequent when the template A and T were on the leading strand, suggesting that lagging-strand synthesis was more error-prone, or less well corrected by proofreading, than was leading strand synthesis.
Keywords: DNA polymerase fidelity; DNA replication accuracy; mismatch repair; mutation accumulation; mutation hotspots.
Copyright © 2018 by the Genetics Society of America.
Figures
References
-
- Bebenek K., Joyce C. M., Fitzgerald M. P., Kunkel T. A., 1990. The fidelity of DNA synthesis catalyzed by derivatives of Escherichia coli DNA polymerase I. J. Biol. Chem. 265: 13878–13887. - PubMed
-
- Bloom L. B., Otto M. R., Eritja R., Reha-Krantz L. J., Goodman M. F., et al. , 1994. Pre-steady-state kinetic analysis of sequence-dependent nucleotide excision by the 3′-exonuclease activity of bacteriophage T4 DNA polymerase. Biochemistry 33: 7576–7586. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
