

A Milieu Molecule for TGF-β Required for Microglia Function in the Nervous System
- PMID: 29909984
- PMCID: PMC6089614
- DOI: 10.1016/j.cell.2018.05.027
A Milieu Molecule for TGF-β Required for Microglia Function in the Nervous System
Abstract
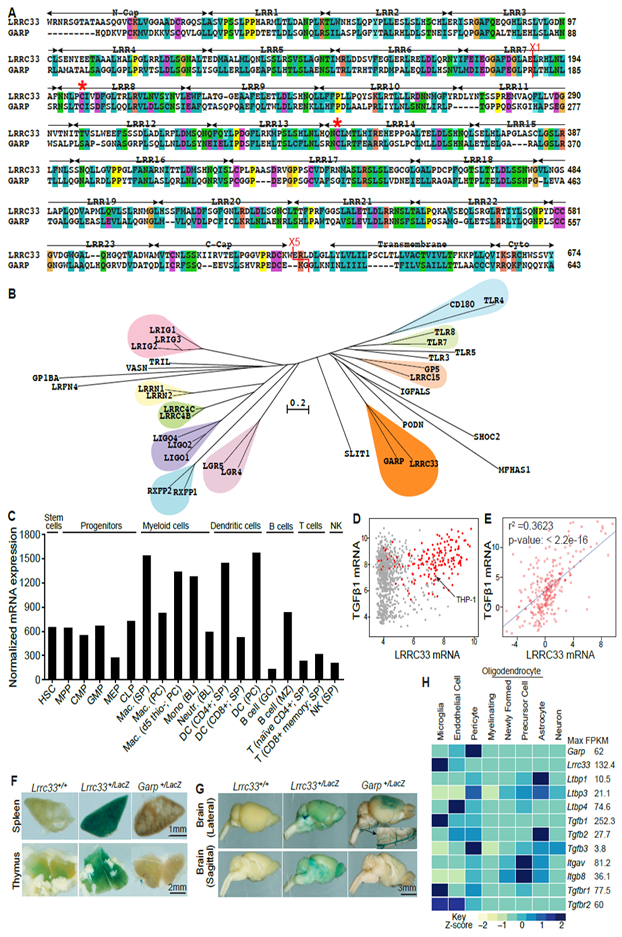
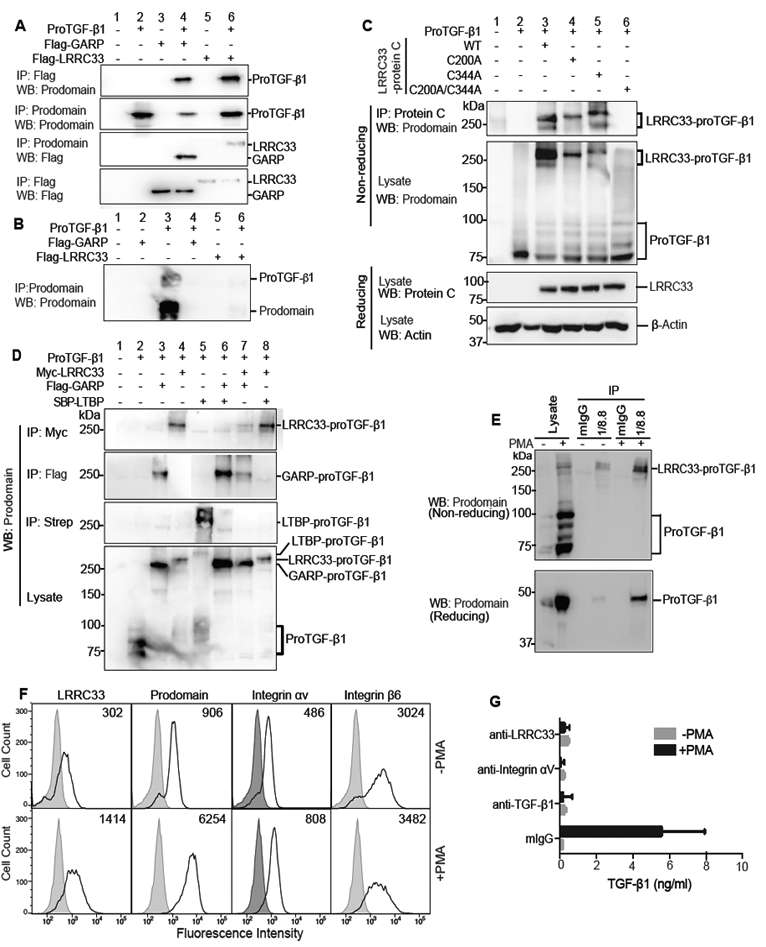
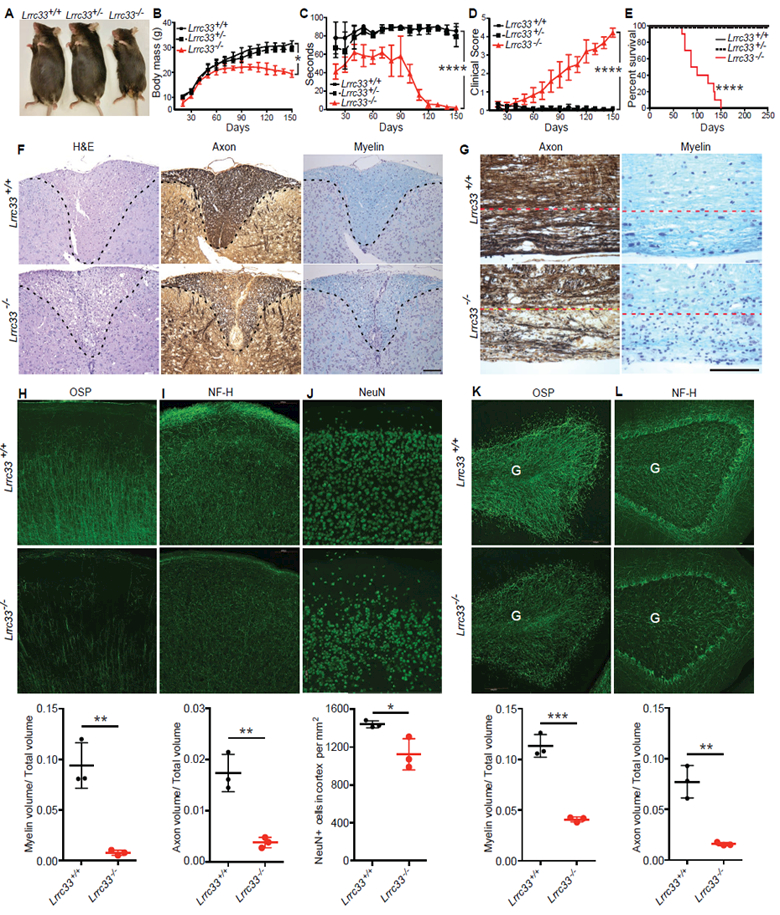
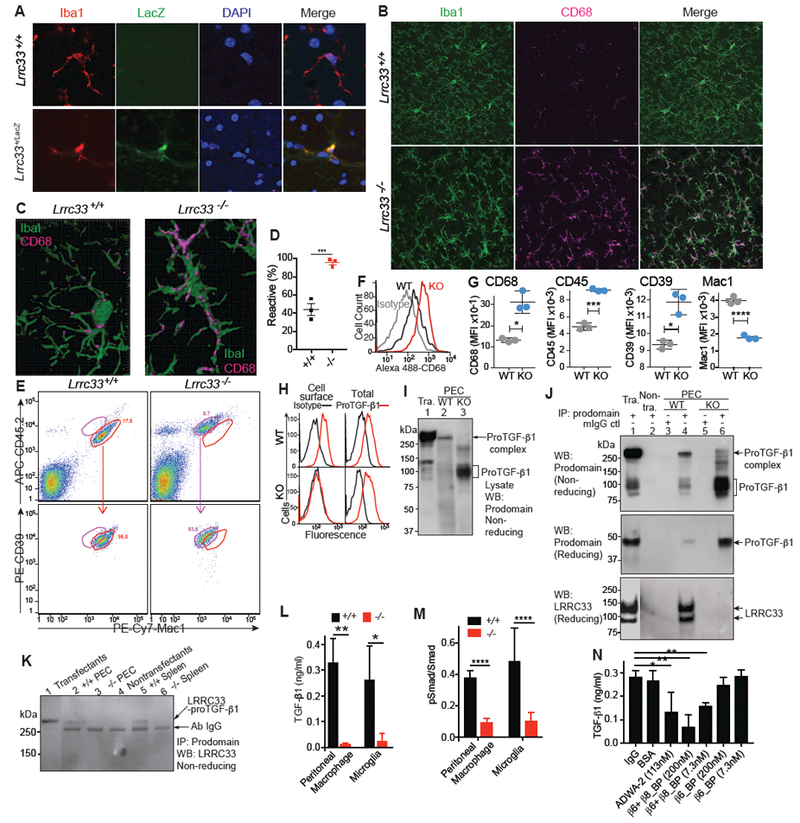
Extracellular proTGF-β is covalently linked to "milieu" molecules in the matrix or on cell surfaces and is latent until TGF-β is released by integrins. Here, we show that LRRC33 on the surface of microglia functions as a milieu molecule and enables highly localized, integrin-αVβ8-dependent TGF-β activation. Lrrc33-/- mice lack CNS vascular abnormalities associated with deficiency in TGF-β-activating integrins but have microglia with a reactive phenotype and after 2 months develop ascending paraparesis with loss of myelinated axons and death by 5 months. Whole bone marrow transplantation results in selective repopulation of Lrrc33-/- brains with WT microglia and halts disease progression. The phenotypes of WT and Lrrc33-/- microglia in the same brain suggest that there is little spreading of TGF-β activated from one microglial cell to neighboring microglia. Our results suggest that interactions between integrin-bearing cells and cells bearing milieu molecule-associated TGF-β provide localized and selective activation of TGF-β.
Keywords: LRRC33; TGF-β; integrins; microglia; milieu molecules.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
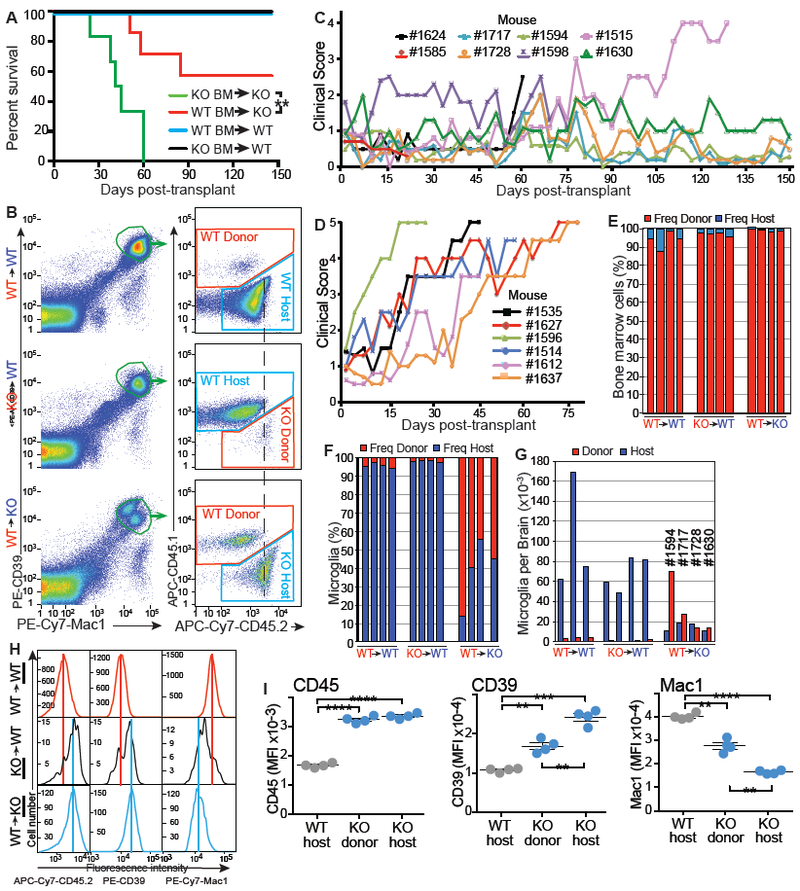
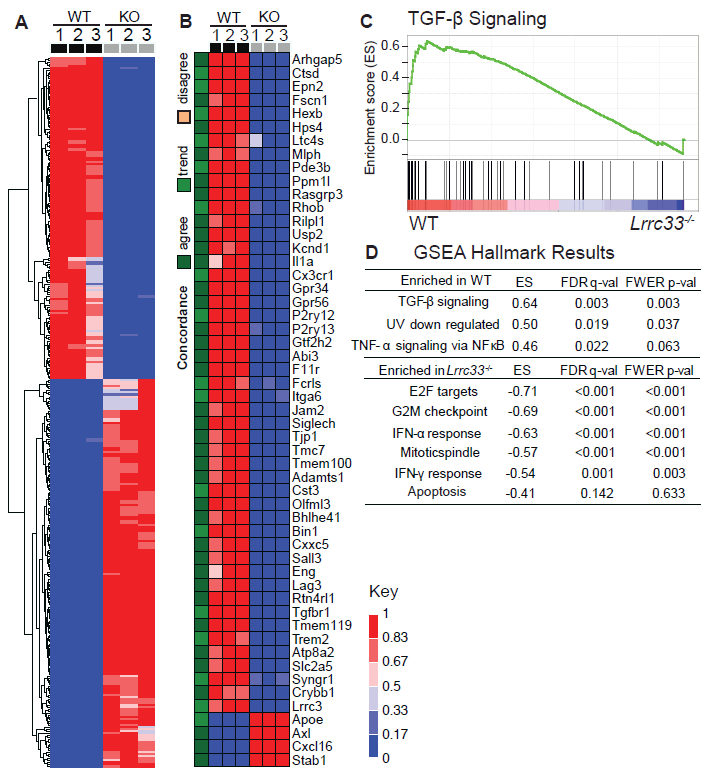
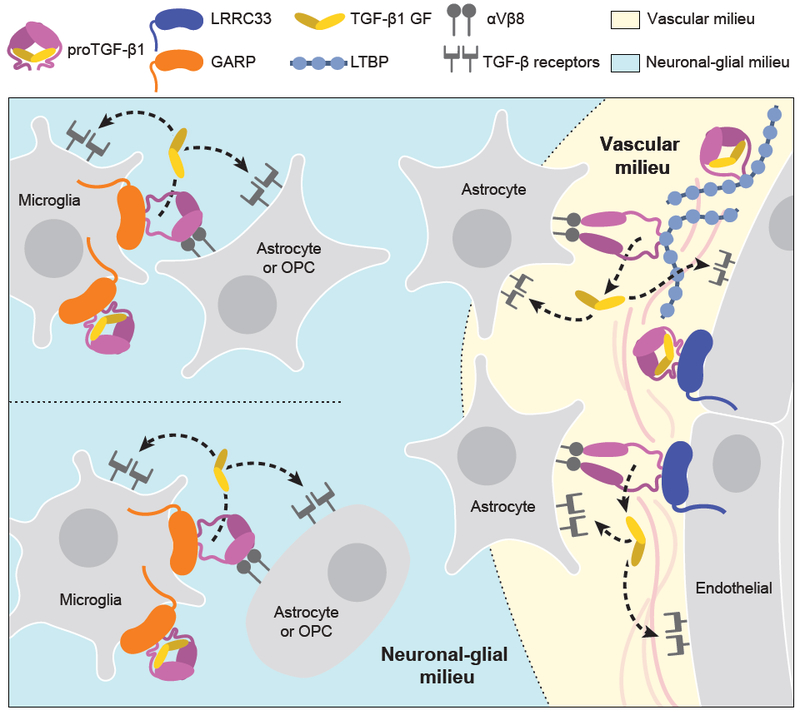
Comment in
-
Lean on Me: Cell-Cell Interactions Release TGF-β for Local Consumption Only.Cell. 2018 Jun 28;174(1):18-20. doi: 10.1016/j.cell.2018.06.008. Cell. 2018. PMID: 29958107
References
-
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, and Rifkin DB (1994). An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem 216, 276–284. - PubMed
-
- Aricescu AR, Lu W, and Jones EY (2006). A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr D Biol Crystallogr 62, 1243–1250. - PubMed
-
- Barash S, Wang W, and Shi Y (2002). Human secretory signal peptide description by hidden Markov model and generation of a strong artificial signal peptide for secreted protein expression. Biochem Biophys Res Commun 294, 835–842. - PubMed
-
- Bilimoria PM, and Stevens B (2015). Microglia function during brain development: New insights from animal models. Brain Res 1617, 7–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
