

A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer
- PMID: 29909987
- PMCID: PMC6046260
- DOI: 10.1016/j.cell.2018.05.037
A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer
Abstract
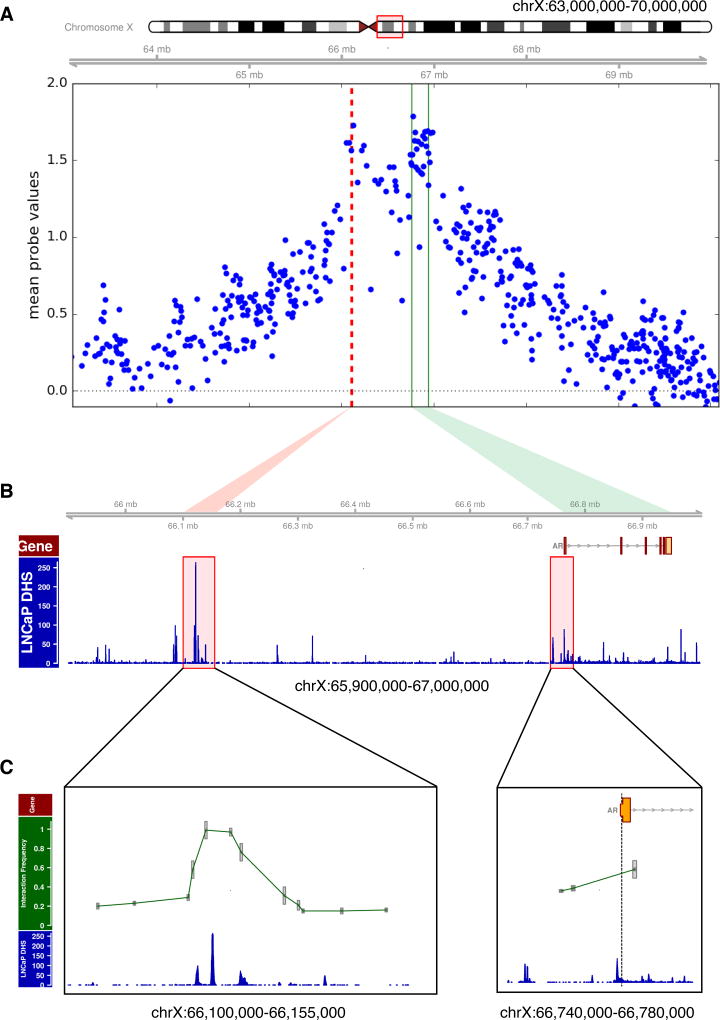
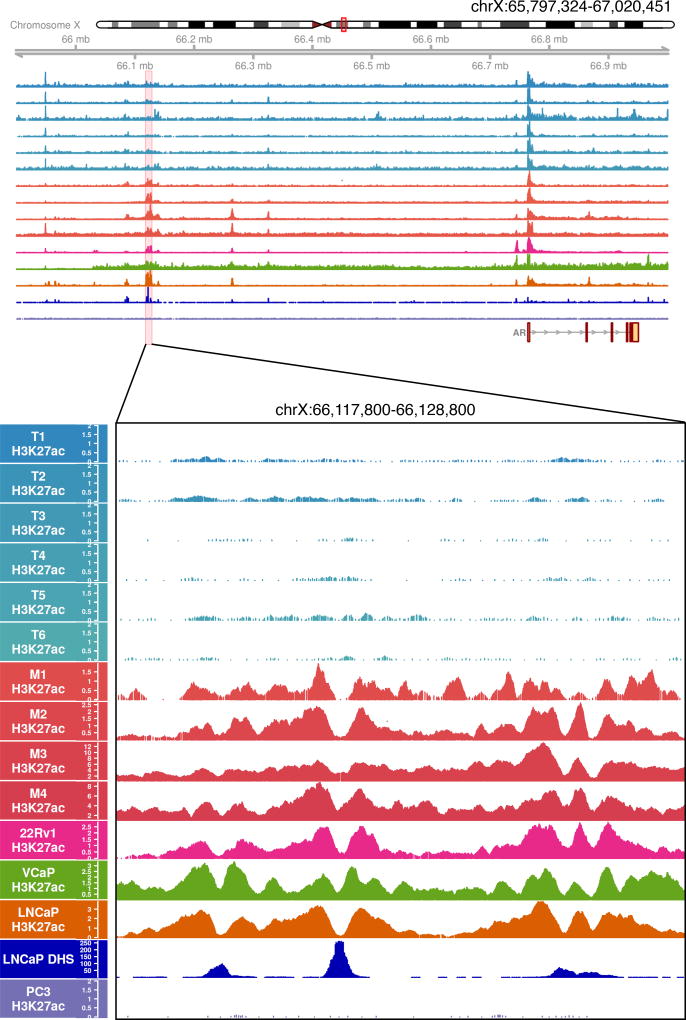
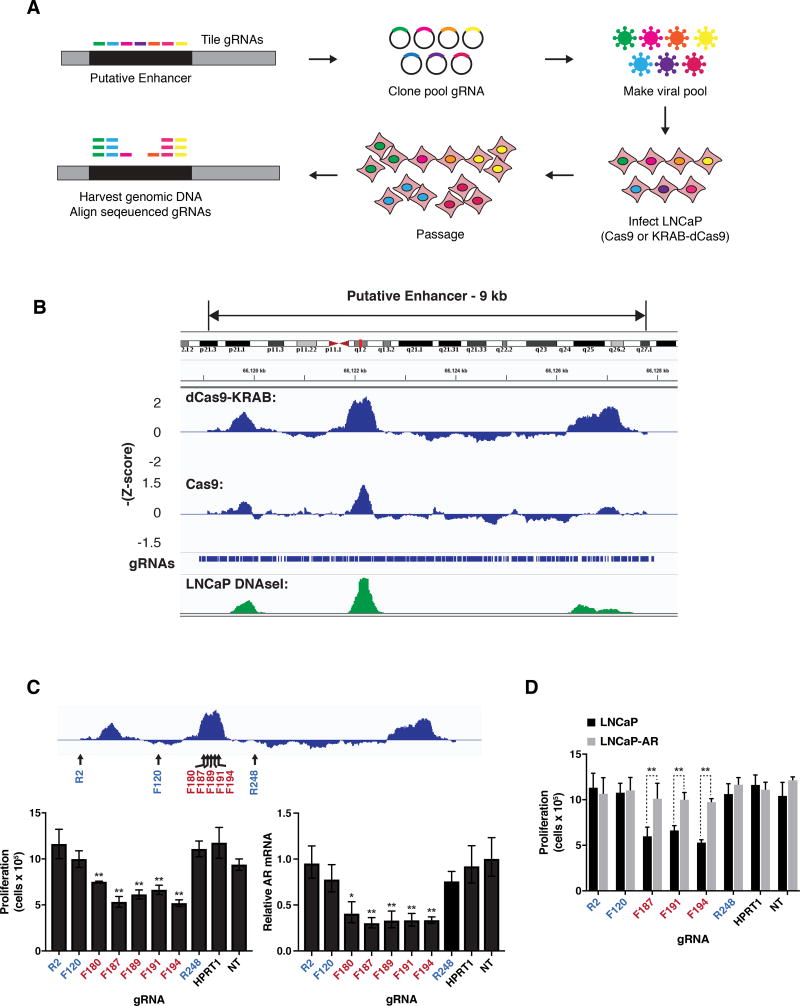
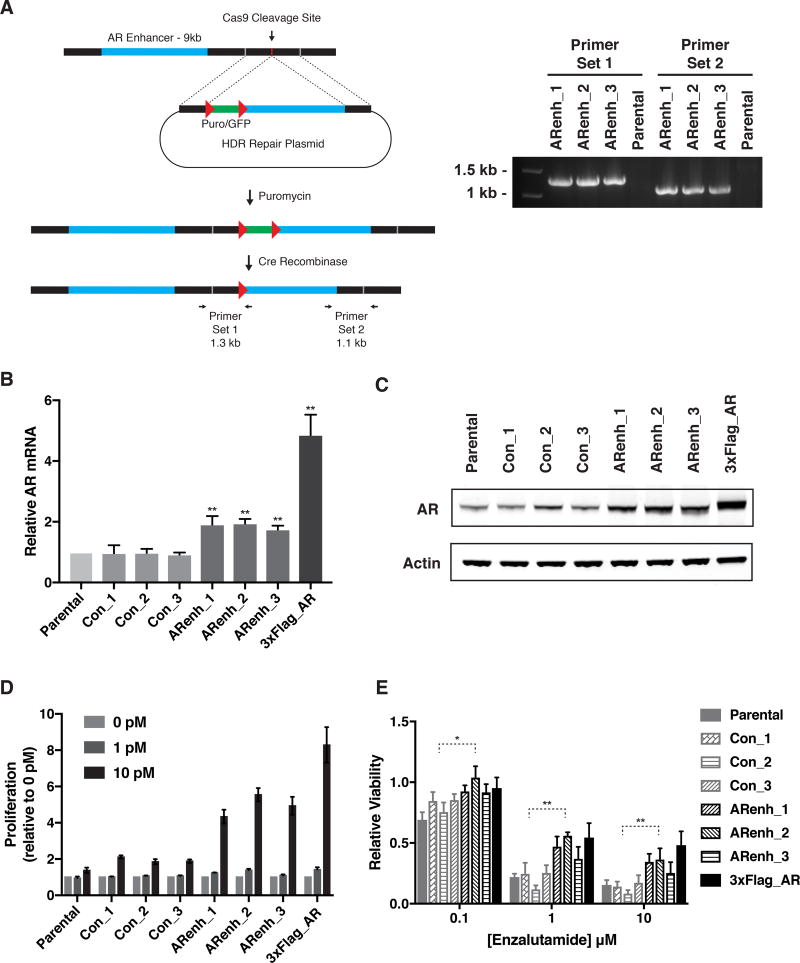
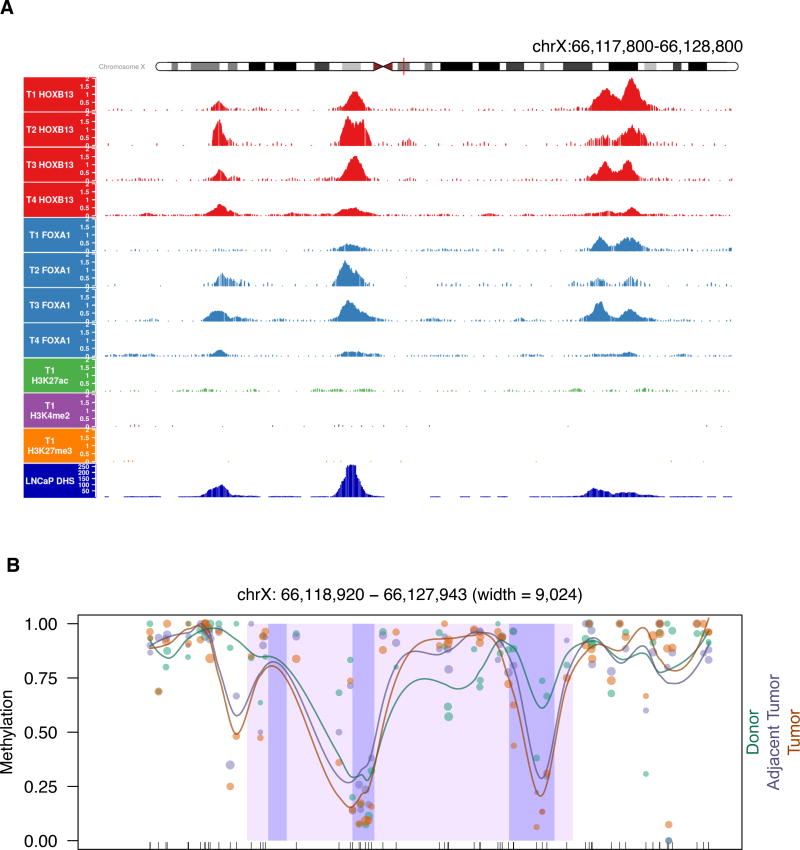
Increased androgen receptor (AR) activity drives therapeutic resistance in advanced prostate cancer. The most common resistance mechanism is amplification of this locus presumably targeting the AR gene. Here, we identify and characterize a somatically acquired AR enhancer located 650 kb centromeric to the AR. Systematic perturbation of this enhancer using genome editing decreased proliferation by suppressing AR levels. Insertion of an additional copy of this region sufficed to increase proliferation under low androgen conditions and to decrease sensitivity to enzalutamide. Epigenetic data generated in localized prostate tumors and benign specimens support the notion that this region is a developmental enhancer. Collectively, these observations underscore the importance of epigenomic profiling in primary specimens and the value of deploying genome editing to functionally characterize noncoding elements. More broadly, this work identifies a therapeutic vulnerability for targeting the AR and emphasizes the importance of regulatory elements as highly recurrent oncogenic drivers.
Keywords: androgen receptor; castrate resistant; enhancer; epigenetic; epigenome editing; epigenomic; functional genomics; genome editing; prostate cancer.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
New genomic drivers identified.Nat Rev Urol. 2018 Sep;15(9):525. doi: 10.1038/s41585-018-0057-2. Nat Rev Urol. 2018. PMID: 29977054 No abstract available.
-
Sequence of events in prostate cancer.Nature. 2018 Aug;560(7720):557-559. doi: 10.1038/d41586-018-06029-5. Nature. 2018. PMID: 30143757 No abstract available.
-
Re: A Somatically Acquired Enhancer of the Androgen Receptor is a Noncoding Driver in Advanced Prostate Cancer.J Urol. 2019 Feb;201(2):230. doi: 10.1097/01.JU.0000553017.28746.79. J Urol. 2019. PMID: 30634342 No abstract available.
References
-
- Blok LJ, Themmen AP, Peters AH, Trapman J, Baarends WM, Hoogerbrugge JW, Grootegoed JA. Transcriptional regulation of androgen receptor gene expression in Sertoli cells and other cell types. Molecular and cellular endocrinology. 1992;88:153–164. - PubMed
-
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nature medicine. 2004;10:33–39. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
