

Circular permutation profiling by deep sequencing libraries created using transposon mutagenesis
- PMID: 29912470
- PMCID: PMC6061844
- DOI: 10.1093/nar/gky255
Circular permutation profiling by deep sequencing libraries created using transposon mutagenesis
Abstract
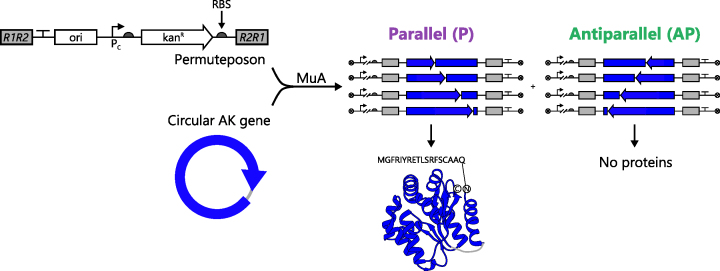
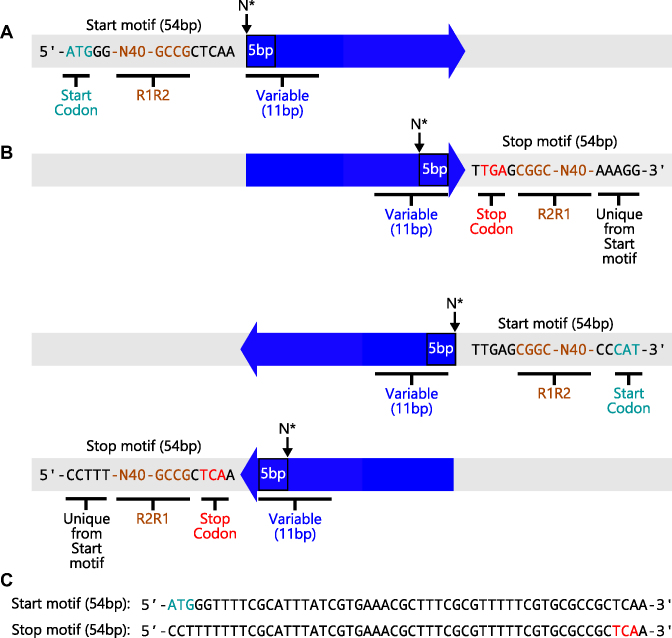
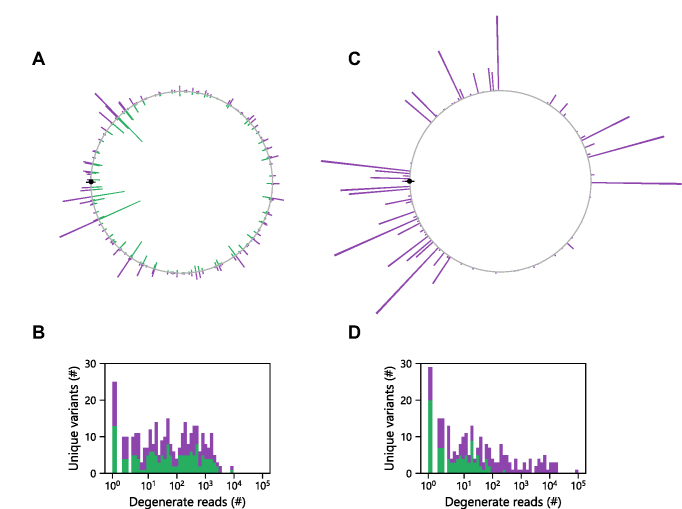
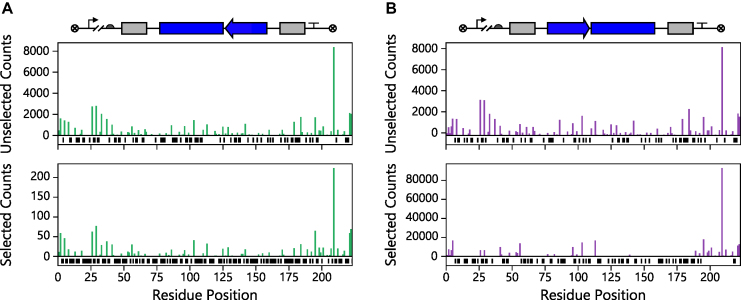
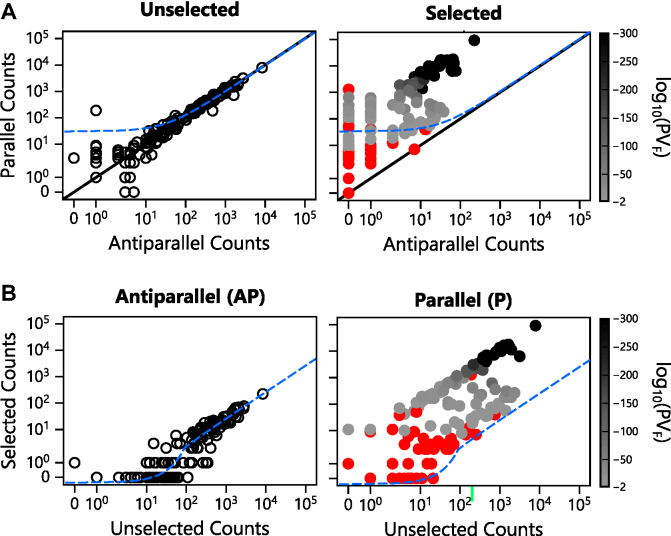
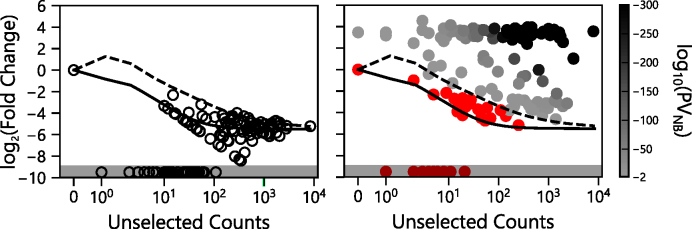
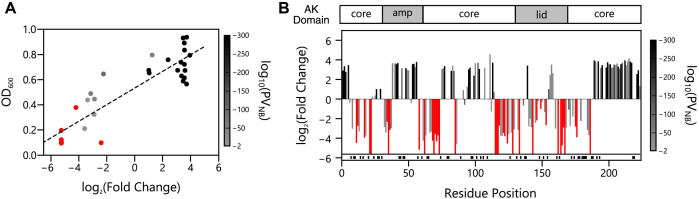
Deep mutational scanning has been used to create high-resolution DNA sequence maps that illustrate the functional consequences of large numbers of point mutations. However, this approach has not yet been applied to libraries of genes created by random circular permutation, an engineering strategy that is used to create open reading frames that express proteins with altered contact order. We describe a new method, termed circular permutation profiling with DNA sequencing (CPP-seq), which combines a one-step transposon mutagenesis protocol for creating libraries with a functional selection, deep sequencing and computational analysis to obtain unbiased insight into a protein's tolerance to circular permutation. Application of this method to an adenylate kinase revealed that CPP-seq creates two types of vectors encoding each circularly permuted gene, which differ in their ability to express proteins. Functional selection of this library revealed that >65% of the sampled vectors that express proteins are enriched relative to those that cannot translate proteins. Mapping enriched sequences onto structure revealed that the mobile AMP binding and rigid core domains display greater tolerance to backbone fragmentation than the mobile lid domain, illustrating how CPP-seq can be used to relate a protein's biophysical characteristics to the retention of activity upon permutation.
Figures
References
-
- Starita L.M., Fields S.. Deep mutational Scanning: a highly parallel method to measure the effects of mutation on protein function. Cold Spring Harb. Protoc. 2015; 2015:711–714. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
