

Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics
- PMID: 29914354
- PMCID: PMC6007078
- DOI: 10.1186/s12864-018-4772-0
Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics
Abstract
Background: Single-cell transcriptomics allows researchers to investigate complex communities of heterogeneous cells. It can be applied to stem cells and their descendants in order to chart the progression from multipotent progenitors to fully differentiated cells. While a variety of statistical and computational methods have been proposed for inferring cell lineages, the problem of accurately characterizing multiple branching lineages remains difficult to solve.
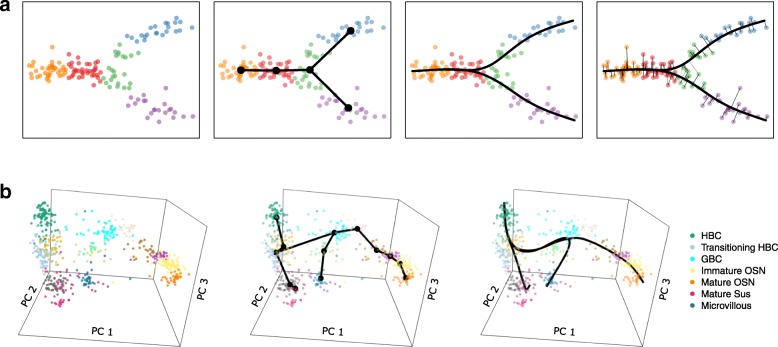
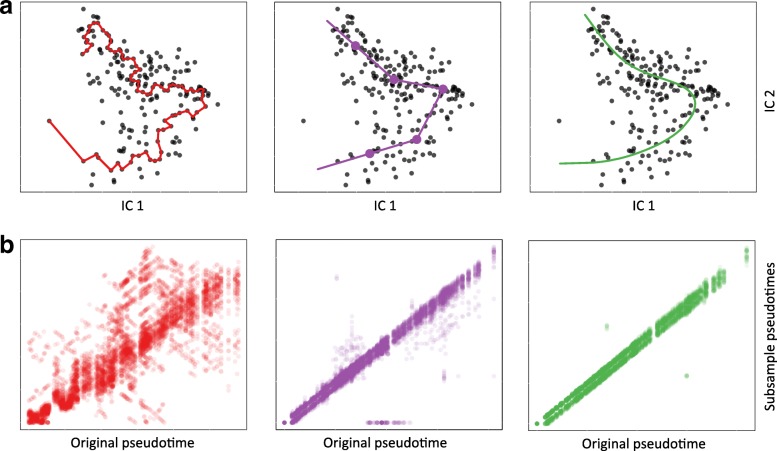
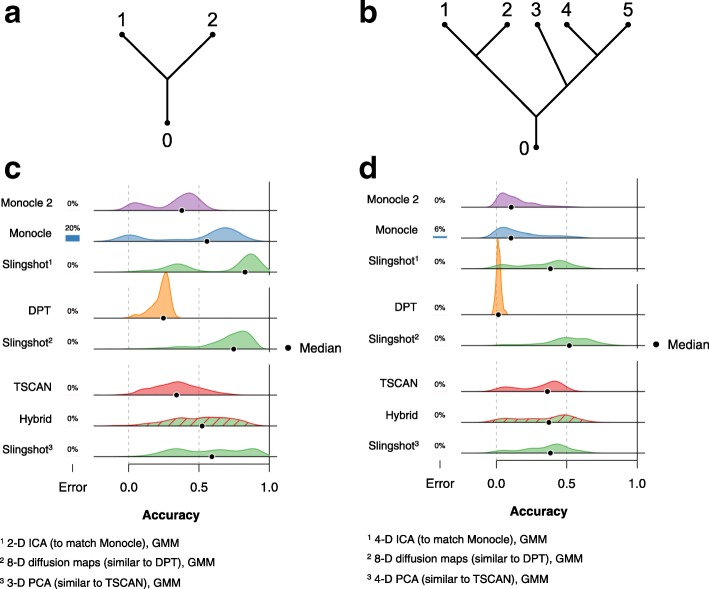
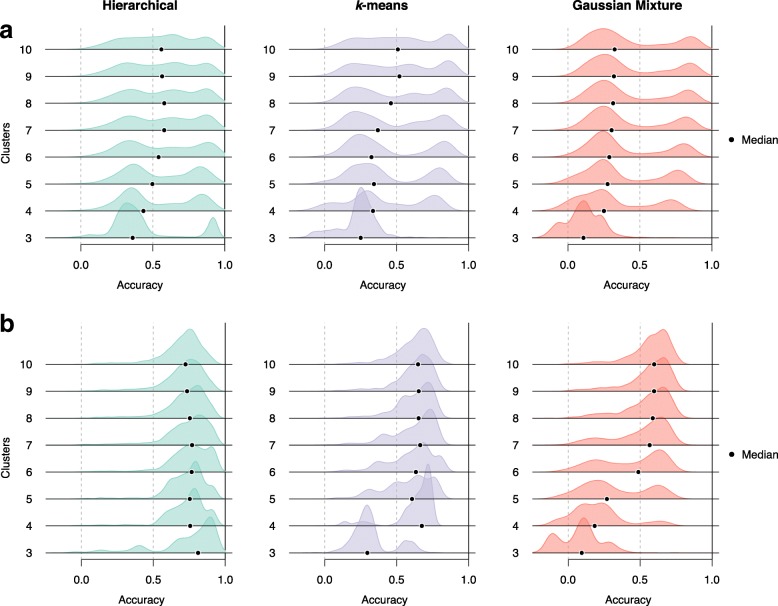
Results: We introduce Slingshot, a novel method for inferring cell lineages and pseudotimes from single-cell gene expression data. In previously published datasets, Slingshot correctly identifies the biological signal for one to three branching trajectories. Additionally, our simulation study shows that Slingshot infers more accurate pseudotimes than other leading methods.
Conclusions: Slingshot is a uniquely robust and flexible tool which combines the highly stable techniques necessary for noisy single-cell data with the ability to identify multiple trajectories. Accurate lineage inference is a critical step in the identification of dynamic temporal gene expression.
Keywords: Lineage inference; Pseudotime inference; RNA-Seq; Single cell.
Conflict of interest statement
Ethics approval and consent to participate
Ethics approval and consent to participate were not applicable to this study.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Campbell K, Ponting CP, Webber C. Laplacian eigenmaps and principal curves for high resolution pseudotemporal ordering of single-cell RNA-seq profiles. Technical report, Functional Genomics Unit, MRC, University of Oxford, UK. 2015. biorxiv.org/content/early/2015/09/18/027219.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
