

Mutations in the SRP54 gene cause severe congenital neutropenia as well as Shwachman-Diamond-like syndrome
- PMID: 29914977
- PMCID: PMC6536700
- DOI: 10.1182/blood-2017-12-820308
Mutations in the SRP54 gene cause severe congenital neutropenia as well as Shwachman-Diamond-like syndrome
Abstract
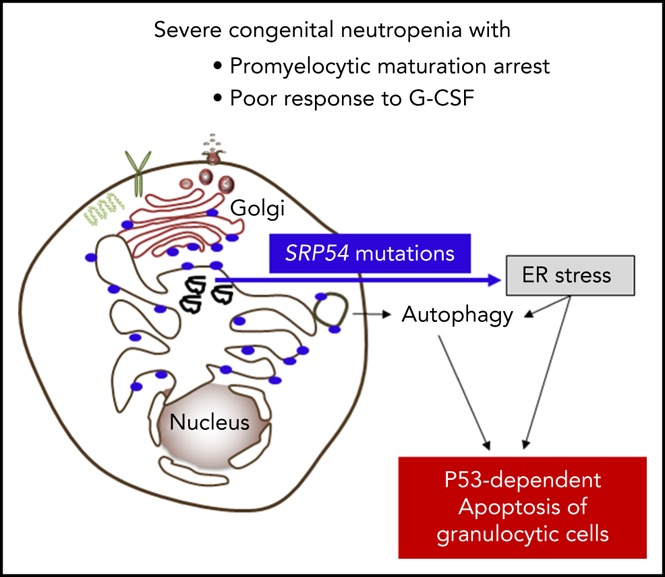
Congenital neutropenias (CNs) are rare heterogeneous genetic disorders, with about 25% of patients without known genetic defects. Using whole-exome sequencing, we identified a heterozygous mutation in the SRP54 gene, encoding the signal recognition particle (SRP) 54 GTPase protein, in 3 sporadic cases and 1 autosomal dominant family. We subsequently sequenced the SRP54 gene in 66 probands from the French CN registry. In total, we identified 23 mutated cases (16 sporadic, 7 familial) with 7 distinct germ line SRP54 mutations including a recurrent in-frame deletion (Thr117del) in 14 cases. In nearly all patients, neutropenia was chronic and profound with promyelocytic maturation arrest, occurring within the first months of life, and required long-term granulocyte colony-stimulating factor therapy with a poor response. Neutropenia was sometimes associated with a severe neurodevelopmental delay (n = 5) and/or an exocrine pancreatic insufficiency requiring enzyme supplementation (n = 3). The SRP54 protein is a key component of the ribonucleoprotein complex that mediates the co-translational targeting of secretory and membrane proteins to the endoplasmic reticulum (ER). We showed that SRP54 was specifically upregulated during the in vitro granulocytic differentiation, and that SRP54 mutations or knockdown led to a drastically reduced proliferation of granulocytic cells associated with an enhanced P53-dependent apoptosis. Bone marrow examination of SRP54-mutated patients revealed a major dysgranulopoiesis and features of cellular ER stress and autophagy that were confirmed using SRP54-mutated primary cells and SRP54 knockdown cells. In conclusion, we characterized a pathological pathway, which represents the second most common cause of CN with maturation arrest in the French CN registry.
© 2018 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures






Comment in
-
SRP54 and a need for a new neutropenia nosology.Blood. 2018 Sep 20;132(12):1220-1222. doi: 10.1182/blood-2018-07-859959. Blood. 2018. PMID: 30237254 Free PMC article.
References
-
- Donadieu J, Beaupain B, Fenneteau O, Bellanné-Chantelot C. Congenital neutropenia in the era of genomics: classification, diagnosis, and natural history. Br J Haematol. 2017;179(4):557-574. - PubMed
-
- Donadieu J, Beaupain B, Mahlaoui N, Bellanné-Chantelot C. Epidemiology of congenital neutropenia. Hematol Oncol Clin North Am. 2013;27(1):1-17, vii. - PubMed
-
- Glaubach T, Minella AC, Corey SJ. Cellular stress pathways in pediatric bone marrow failure syndromes: many roads lead to neutropenia. Pediatr Res. 2014;75(1-2):189-195. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
