

Enantioselective remote meta-C-H arylation and alkylation via a chiral transient mediator
- PMID: 29915312
- PMCID: PMC6026055
- DOI: 10.1038/s41586-018-0220-1
Enantioselective remote meta-C-H arylation and alkylation via a chiral transient mediator
Abstract
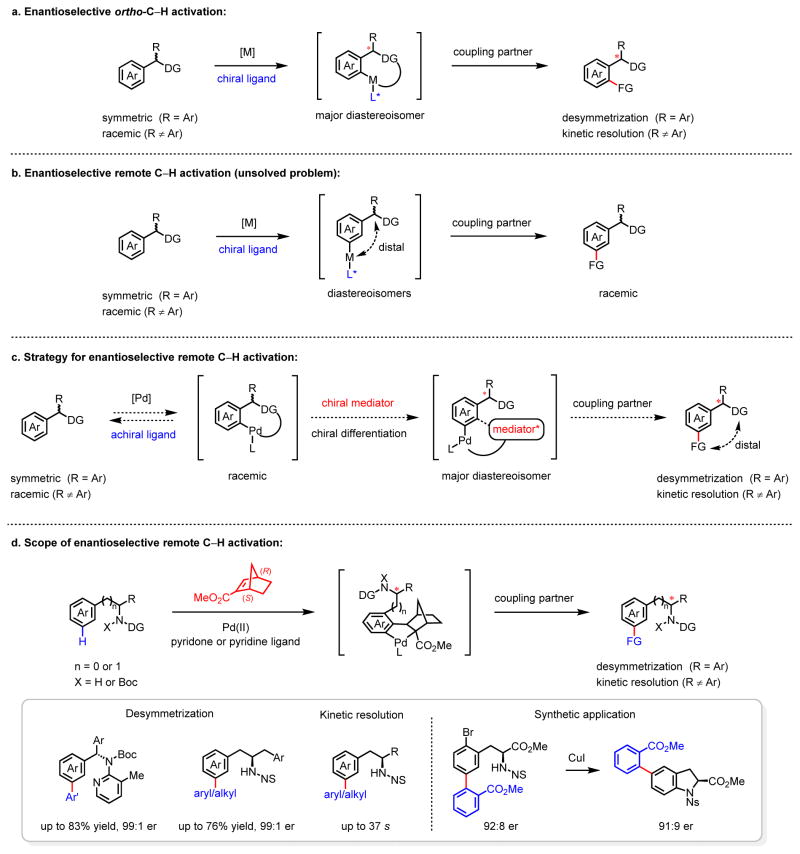
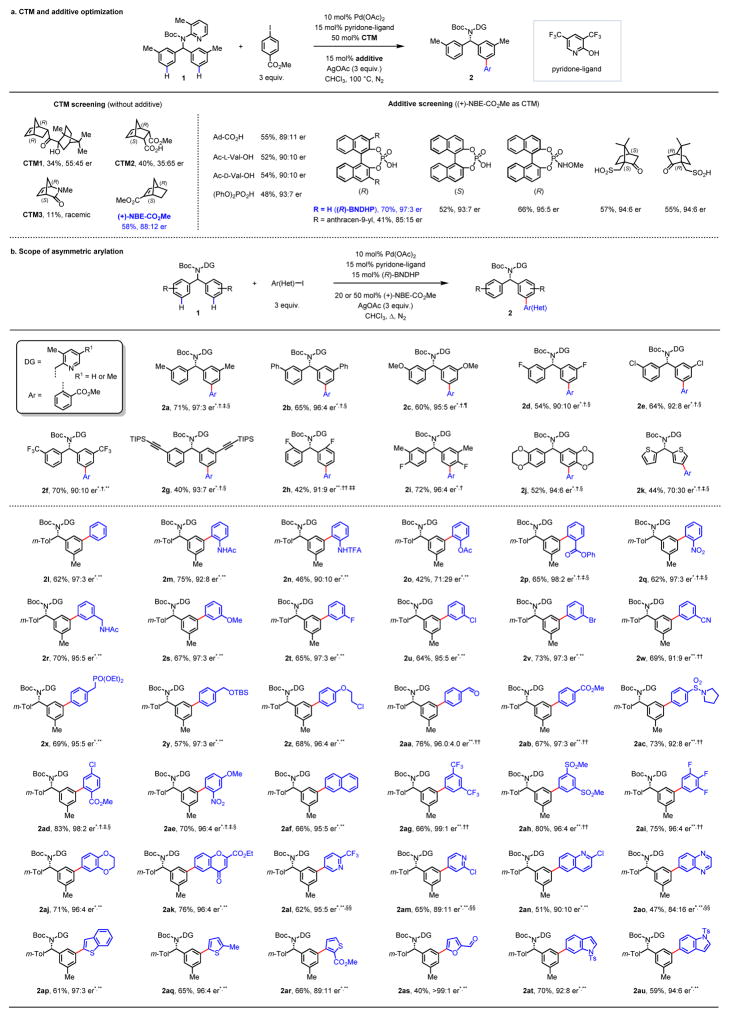
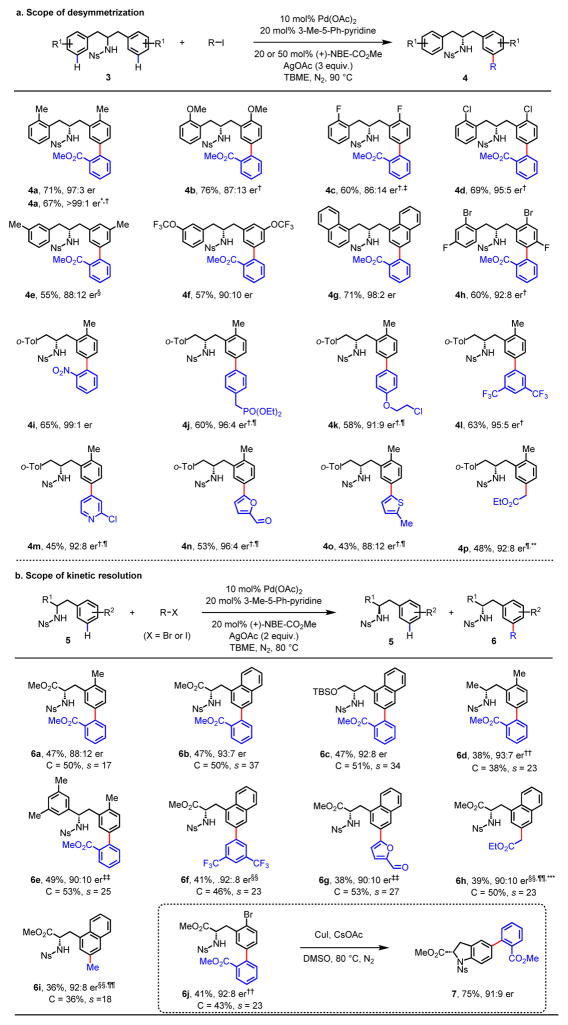
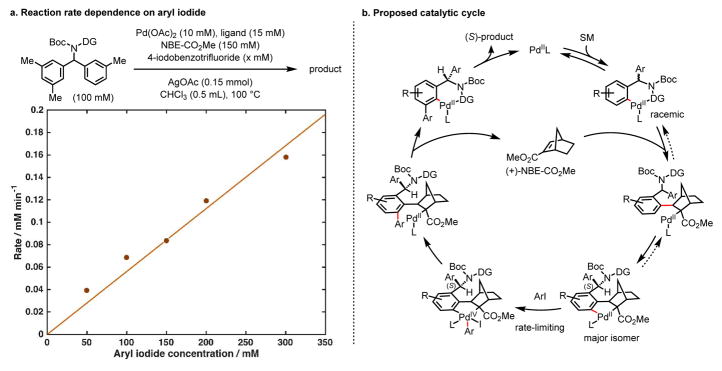
Enantioselective carbon-hydrogen (C-H) activation reactions by asymmetric metallation could provide new routes for the construction of chiral molecules1,2. However, current methods are typically limited to the formation of five- or six-membered metallacycles, thereby preventing the asymmetric functionalization of C-H bonds at positions remote to existing functional groups. Here we report enantioselective remote C-H activation using a catalytic amount of a chiral norbornene as a transient mediator, which relays initial ortho-C-H activation to the meta position. This was used in the enantioselective meta-C-H arylation of benzylamines, as well as the arylation and alkylation of homobenzylamines. The enantioselectivities obtained using the chiral transient mediator are comparable across different classes of substrates containing either neutral σ-donor or anionic coordinating groups. This relay strategy could provide an alternative means to remote chiral induction, one of the most challenging problems in asymmetric catalysis3,4.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Ligand-enabled meta-C-H activation using a transient mediator.Nature. 2015 Mar 19;519(7543):334-8. doi: 10.1038/nature14214. Epub 2015 Mar 9. Nature. 2015. PMID: 25754328 Free PMC article.
-
Differentiation and functionalization of remote C-H bonds in adjacent positions.Nat Chem. 2020 Apr;12(4):399-404. doi: 10.1038/s41557-020-0424-5. Epub 2020 Mar 2. Nat Chem. 2020. PMID: 32123338 Free PMC article.
-
Pd(II)-catalyzed regioselective 2-alkylation of indoles via a norbornene-mediated C-H activation: mechanism and applications.J Am Chem Soc. 2012 Sep 5;134(35):14563-72. doi: 10.1021/ja3058138. Epub 2012 Aug 22. J Am Chem Soc. 2012. PMID: 22913367
-
Enantioselective formation of quaternary carbon stereocenters in natural product synthesis: a recent update.Nat Prod Rep. 2020 Feb 26;37(2):276-292. doi: 10.1039/c9np00039a. Nat Prod Rep. 2020. PMID: 31515549 Review.
-
Catalytic C-H functionalization by metalloporphyrins: recent developments and future directions.Chem Soc Rev. 2011 Apr;40(4):1899-909. doi: 10.1039/c0cs00070a. Epub 2010 Nov 19. Chem Soc Rev. 2011. PMID: 21088785 Review.
Cited by
-
RhodiumIII-catalyzed remote difunctionalization of arenes assisted by a relay directing group.Chem Sci. 2022 May 30;13(24):7347-7354. doi: 10.1039/d2sc02205b. eCollection 2022 Jun 22. Chem Sci. 2022. PMID: 35799802 Free PMC article.
-
Palladium-Catalyzed Stitching of 1,3-C(sp3)-H Bonds with Dihaloarenes: Short Synthesis of (±)-Echinolactone D.J Am Chem Soc. 2023 Aug 16;145(32):17919-17925. doi: 10.1021/jacs.3c05383. Epub 2023 Aug 1. J Am Chem Soc. 2023. PMID: 37526629 Free PMC article.
-
Enantioselective transition metal catalysis directed by chiral cations.Chem Sci. 2020 Dec 21;12(2):533-539. doi: 10.1039/d0sc05734g. Chem Sci. 2020. PMID: 34163782 Free PMC article. Review.
-
Unlocking regioselective meta-alkylation with epoxides and oxetanes via dynamic kinetic catalyst control.Nat Commun. 2024 Jan 2;15(1):31. doi: 10.1038/s41467-023-44219-6. Nat Commun. 2024. PMID: 38167324 Free PMC article.
-
Enantioselective benzylic C-H arylation via photoredox and nickel dual catalysis.Nat Commun. 2019 Aug 7;10(1):3549. doi: 10.1038/s41467-019-11392-6. Nat Commun. 2019. PMID: 31391466 Free PMC article.
References
-
- Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Transition metal-catalyzed C–H activation reactions: diastereoselectivity and enantioselectivity. Chem Soc Rev. 2009;38:3242–3272. - PubMed
-
- Newton CG, Wang SG, Oliveira CC, Cramer N. Catalytic enantioselective transformations involving C–H bond cleavage by transition-metal complexes. Chem Rev. 2017;117:8908–8976. - PubMed
-
- Kakiuchi F, Gendre PL, Yamada A, Ohtaki H, Murai S. Atropselective alkylation of biaryl compounds by means of transition metal-catalyzed C–H/olefin coupling. Tetrahedron: Asymmetry. 2000;11:2647–2651.
-
- Shi BF, Maugel N, Zhang YH, Yu JQ. PdII-catalyzed enantioselective activation of C(sp2)–H and C(sp3)–H bonds using monoprotected amino acids as chiral ligands. Angew Chem Int Ed. 2008;47:4882–4886. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
