

Oncogenic TRK fusions are amenable to inhibition in hematologic malignancies
- PMID: 29920189
- PMCID: PMC6118587
- DOI: 10.1172/JCI120787
Oncogenic TRK fusions are amenable to inhibition in hematologic malignancies
Abstract
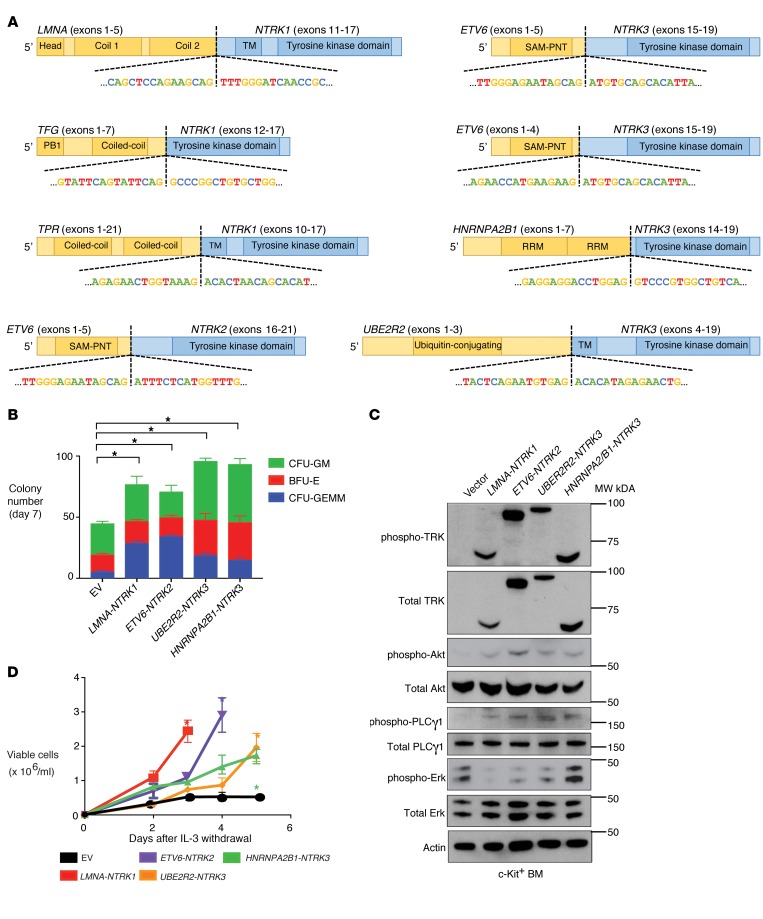
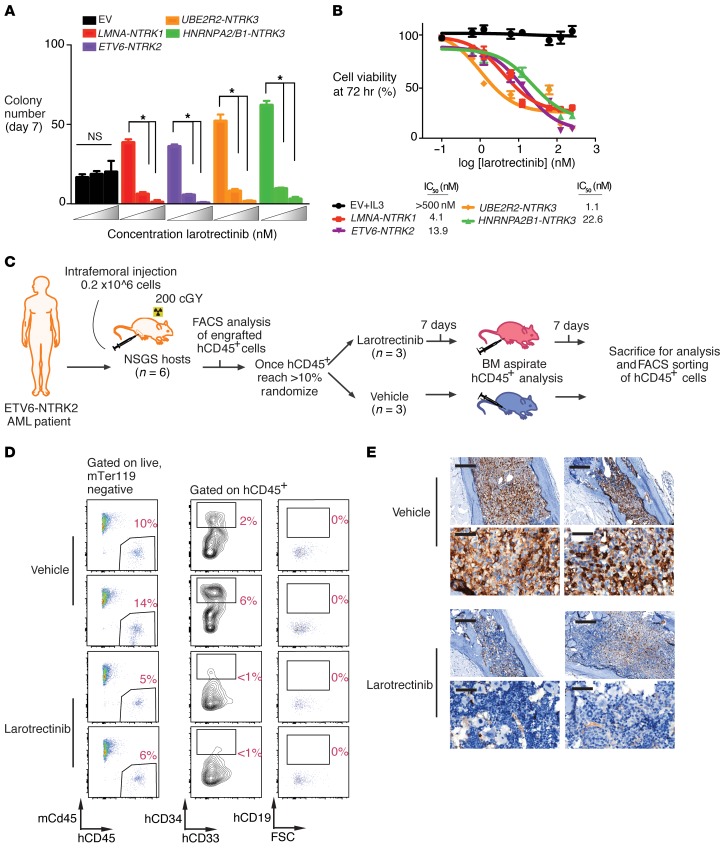
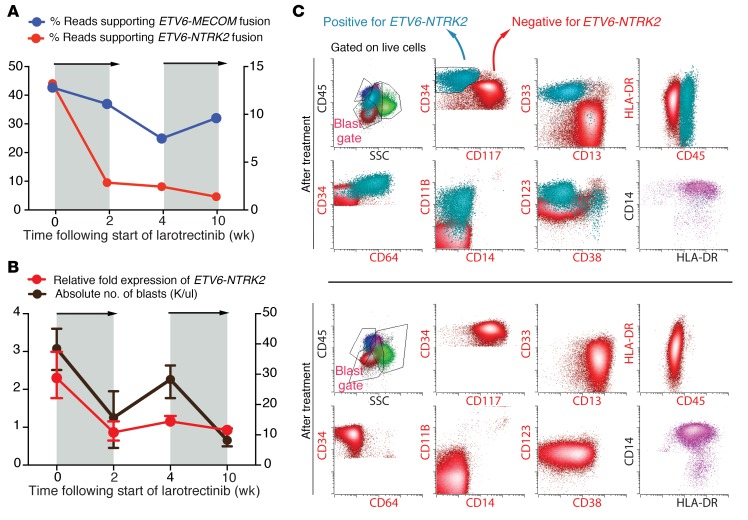
Rearrangements involving the neurotrophic receptor kinase genes (NTRK1, NTRK2, and NTRK3; hereafter referred to as TRK) produce oncogenic fusions in a wide variety of cancers in adults and children. Although TRK fusions occur in fewer than 1% of all solid tumors, inhibition of TRK results in profound therapeutic responses, resulting in Breakthrough Therapy FDA approval of the TRK inhibitor larotrectinib for adult and pediatric patients with solid tumors, regardless of histology. In contrast to solid tumors, the frequency of TRK fusions and the clinical effects of targeting TRK in hematologic malignancies are unknown. Here, through an evaluation for TRK fusions across more than 7,000 patients with hematologic malignancies, we identified TRK fusions in acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), histiocytosis, multiple myeloma, and dendritic cell neoplasms. Although TRK fusions occurred in only 0.1% of patients (8 of 7,311 patients), they conferred responsiveness to TRK inhibition in vitro and in vivo in a patient-derived xenograft and a corresponding AML patient with ETV6-NTRK2 fusion. These data identify that despite their individual rarity, collectively, TRK fusions are present in a wide variety of hematologic malignancies and predict clinically significant therapeutic responses to TRK inhibition.
Keywords: Cancer; Hematology; Oncogenes; Oncology; Signal transduction.
Conflict of interest statement
Figures
References
-
- Skálová A, et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am J Surg Pathol. 2010;34(5):599–608. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
