

dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments
- PMID: 29921301
- PMCID: PMC6010209
- DOI: 10.1186/s13059-018-1449-6
dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments
Abstract
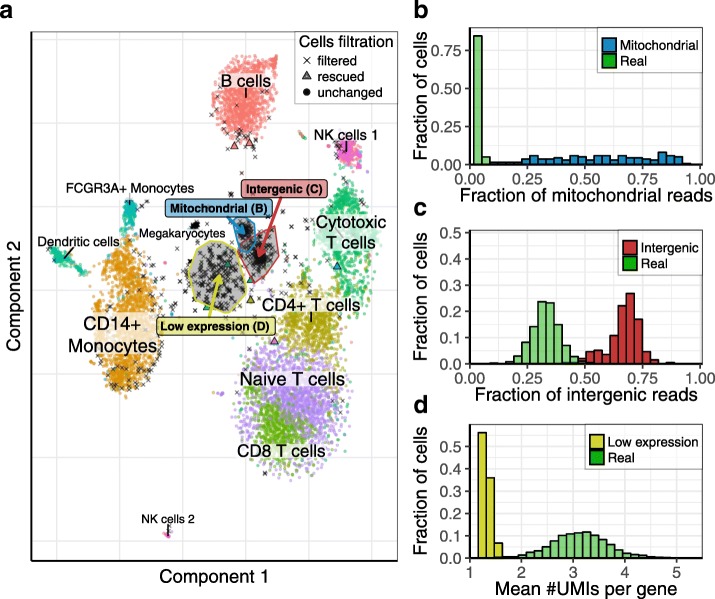
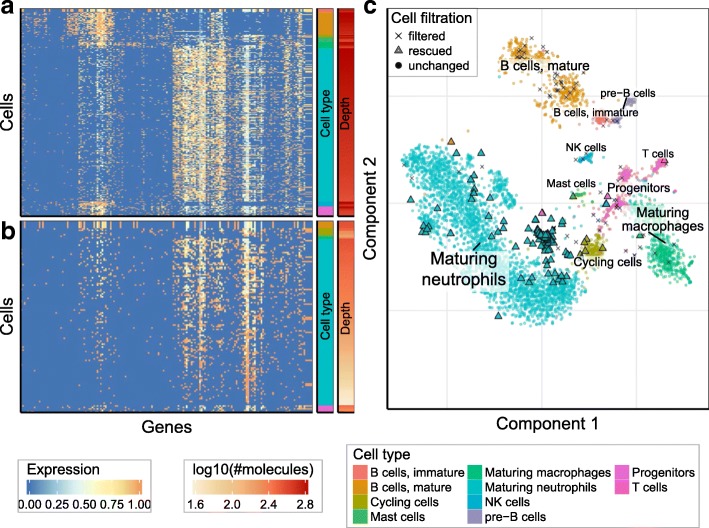
Recent single-cell RNA-seq protocols based on droplet microfluidics use massively multiplexed barcoding to enable simultaneous measurements of transcriptomes for thousands of individual cells. The increasing complexity of such data creates challenges for subsequent computational processing and troubleshooting of these experiments, with few software options currently available. Here, we describe a flexible pipeline for processing droplet-based transcriptome data that implements barcode corrections, classification of cell quality, and diagnostic information about the droplet libraries. We introduce advanced methods for correcting composition bias and sequencing errors affecting cellular and molecular barcodes to provide more accurate estimates of molecular counts in individual cells.
Conflict of interest statement
Ethics approval
The animal work conducted for this study was approved by the Institutional Animal Care and Use Committee (IACUC) of Massachusetts General Hospital.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
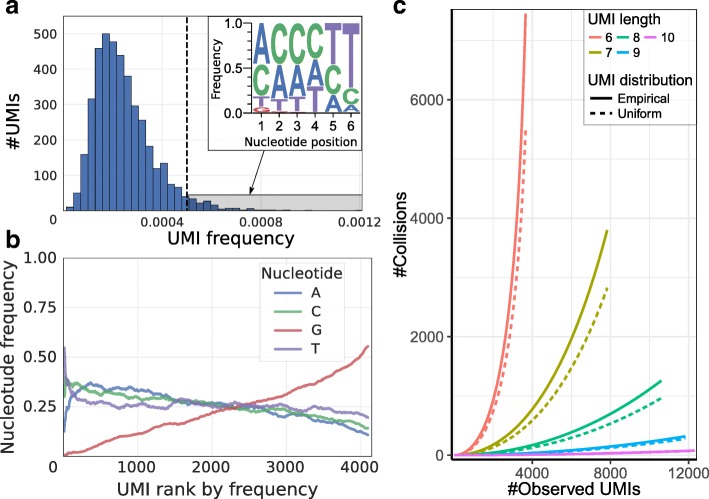
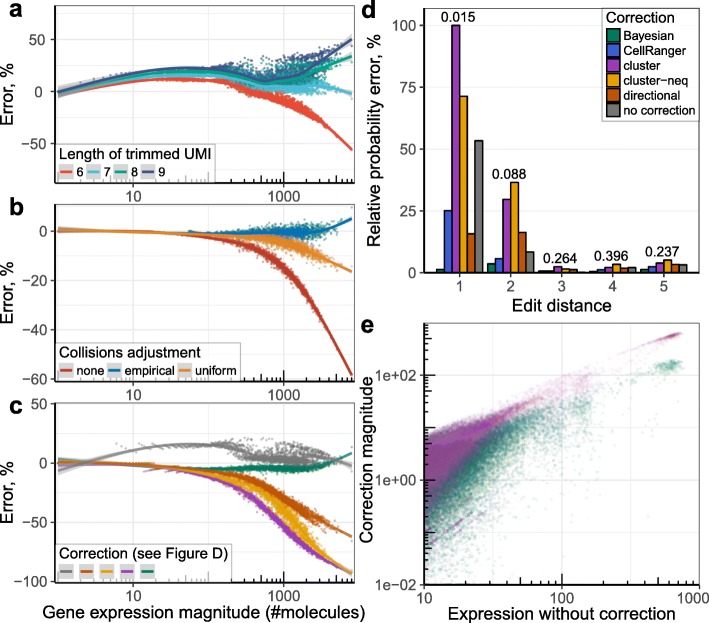
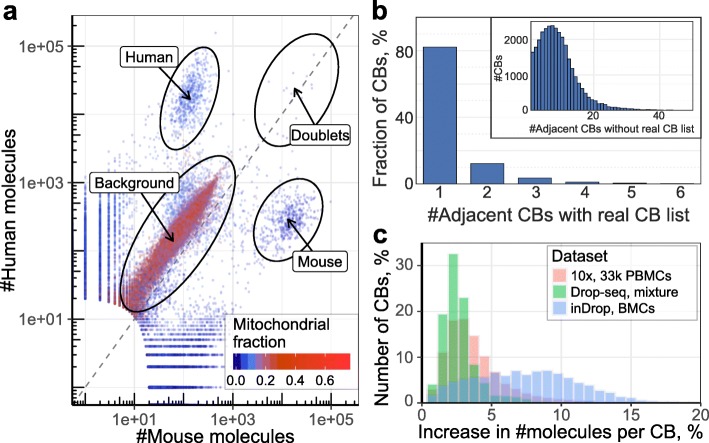
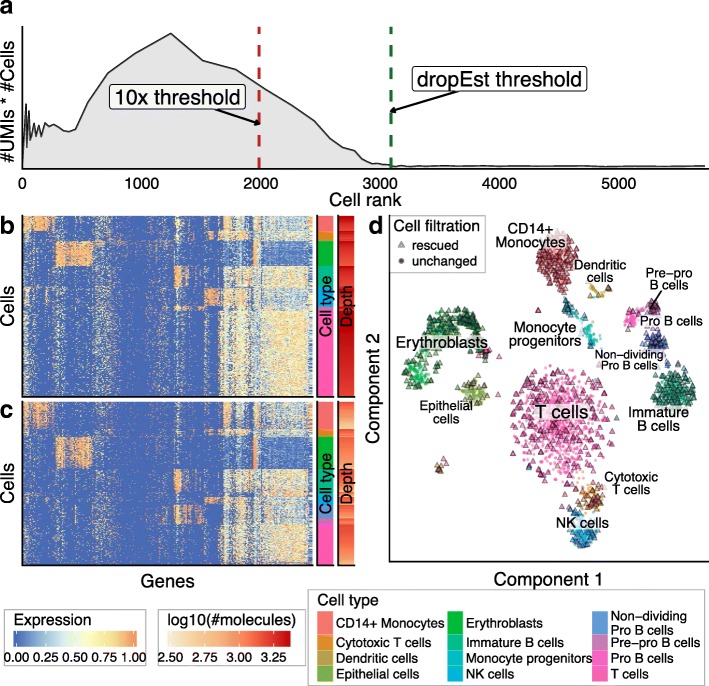
Similar articles
-
Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems.Mol Cell. 2019 Jan 3;73(1):130-142.e5. doi: 10.1016/j.molcel.2018.10.020. Epub 2018 Nov 21. Mol Cell. 2019. PMID: 30472192
-
SCITO-seq: single-cell combinatorial indexed cytometry sequencing.Nat Methods. 2021 Aug;18(8):903-911. doi: 10.1038/s41592-021-01222-3. Epub 2021 Aug 5. Nat Methods. 2021. PMID: 34354295 Free PMC article.
-
Single-Cell Sequencing of 3' RNA Transcripts.Methods Mol Biol. 2024;2822:227-243. doi: 10.1007/978-1-0716-3918-4_16. Methods Mol Biol. 2024. PMID: 38907922
-
The promise of single-cell RNA sequencing for kidney disease investigation.Kidney Int. 2017 Dec;92(6):1334-1342. doi: 10.1016/j.kint.2017.06.033. Epub 2017 Oct 12. Kidney Int. 2017. PMID: 28893418 Free PMC article. Review.
-
Multiplexing Methods for Simultaneous Large-Scale Transcriptomic Profiling of Samples at Single-Cell Resolution.Adv Sci (Weinh). 2021 Sep;8(17):e2101229. doi: 10.1002/advs.202101229. Epub 2021 Jul 8. Adv Sci (Weinh). 2021. PMID: 34240574 Free PMC article. Review.
Cited by
-
Distinct evolutionary paths in chronic lymphocytic leukemia during resistance to the graft-versus-leukemia effect.Sci Transl Med. 2020 Sep 16;12(561):eabb7661. doi: 10.1126/scitranslmed.abb7661. Sci Transl Med. 2020. PMID: 32938797 Free PMC article.
-
Harnessing Single-Cell RNA Sequencing to Identify Dendritic Cell Types, Characterize Their Biological States, and Infer Their Activation Trajectory.Methods Mol Biol. 2023;2618:319-373. doi: 10.1007/978-1-0716-2938-3_22. Methods Mol Biol. 2023. PMID: 36905526
-
Challenges in unsupervised clustering of single-cell RNA-seq data.Nat Rev Genet. 2019 May;20(5):273-282. doi: 10.1038/s41576-018-0088-9. Nat Rev Genet. 2019. PMID: 30617341 Review.
-
Accurate estimation of molecular counts from amplicon sequence data with unique molecular identifiers.Bioinformatics. 2023 Jan 1;39(1):btad002. doi: 10.1093/bioinformatics/btad002. Bioinformatics. 2023. PMID: 36610988 Free PMC article.
-
Characterization of hormone-producing cell types in the teleost pituitary gland using single-cell RNA-seq.Sci Data. 2021 Oct 28;8(1):279. doi: 10.1038/s41597-021-01058-8. Sci Data. 2021. PMID: 34711832 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
