

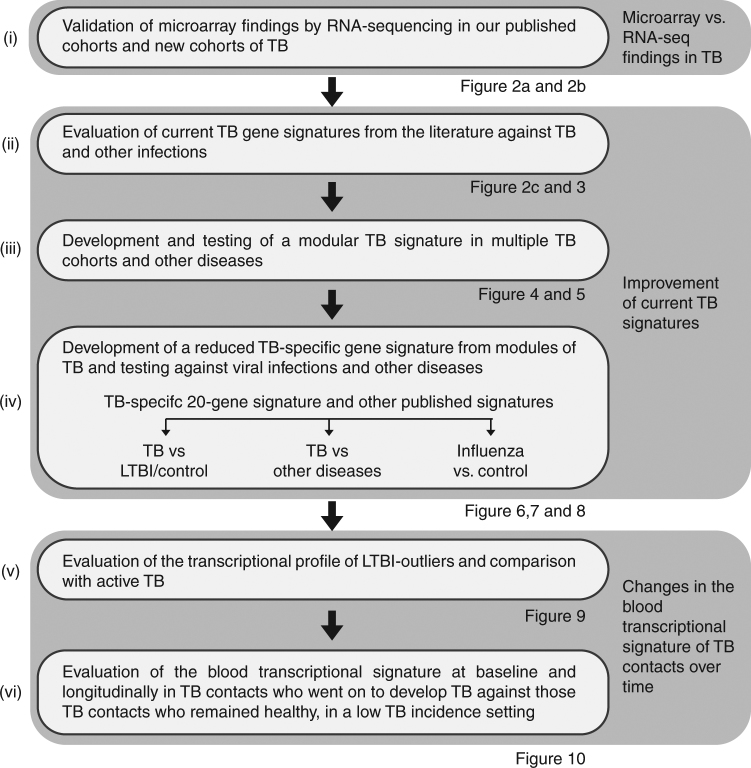
A modular transcriptional signature identifies phenotypic heterogeneity of human tuberculosis infection
- PMID: 29921861
- PMCID: PMC6008327
- DOI: 10.1038/s41467-018-04579-w
A modular transcriptional signature identifies phenotypic heterogeneity of human tuberculosis infection
Abstract
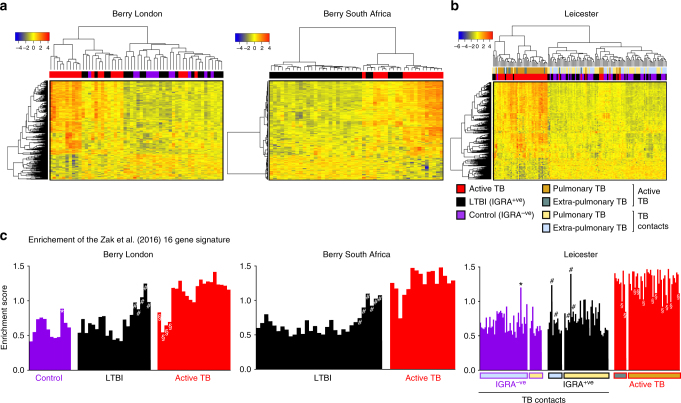
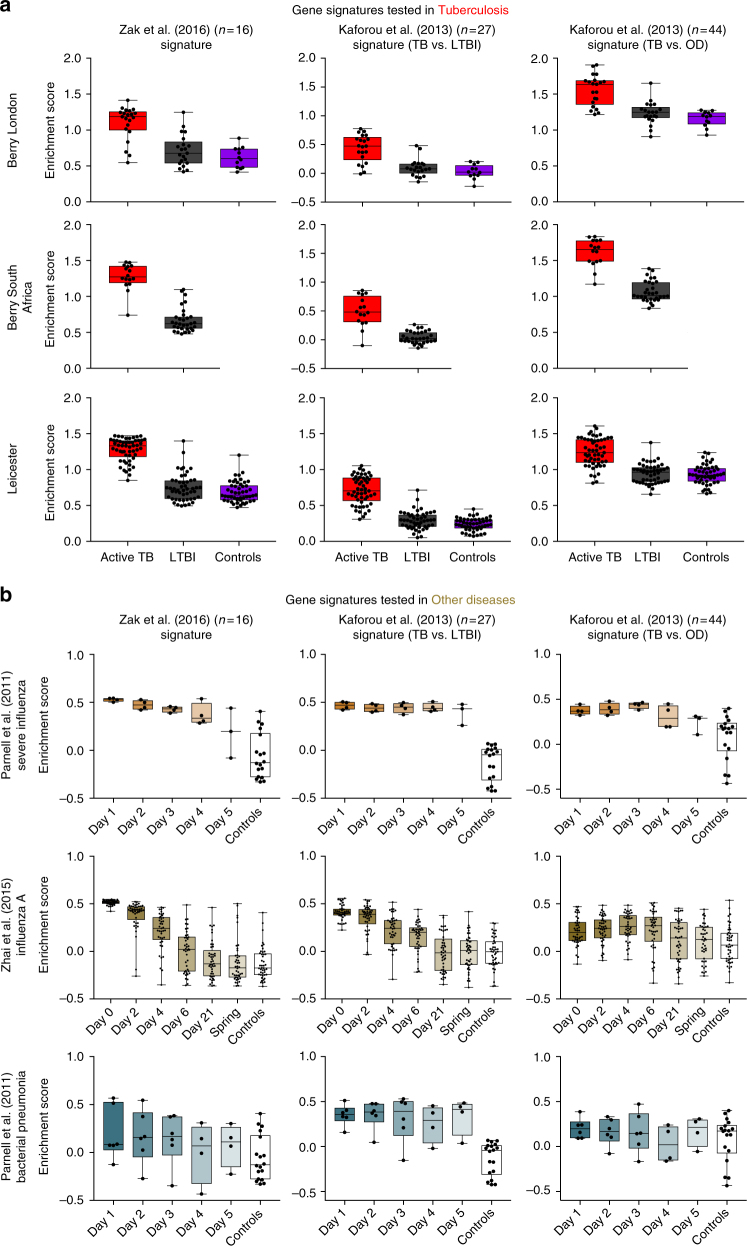
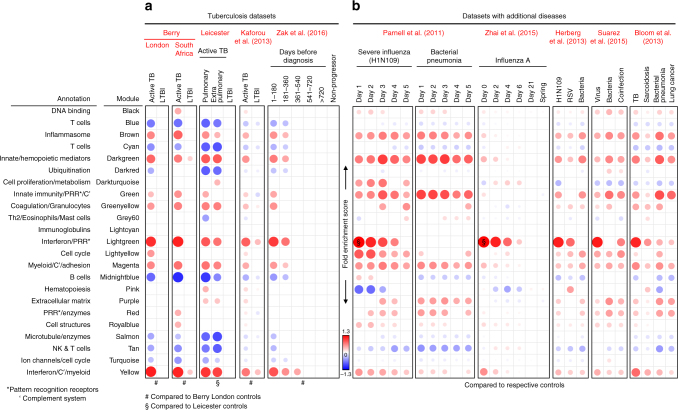
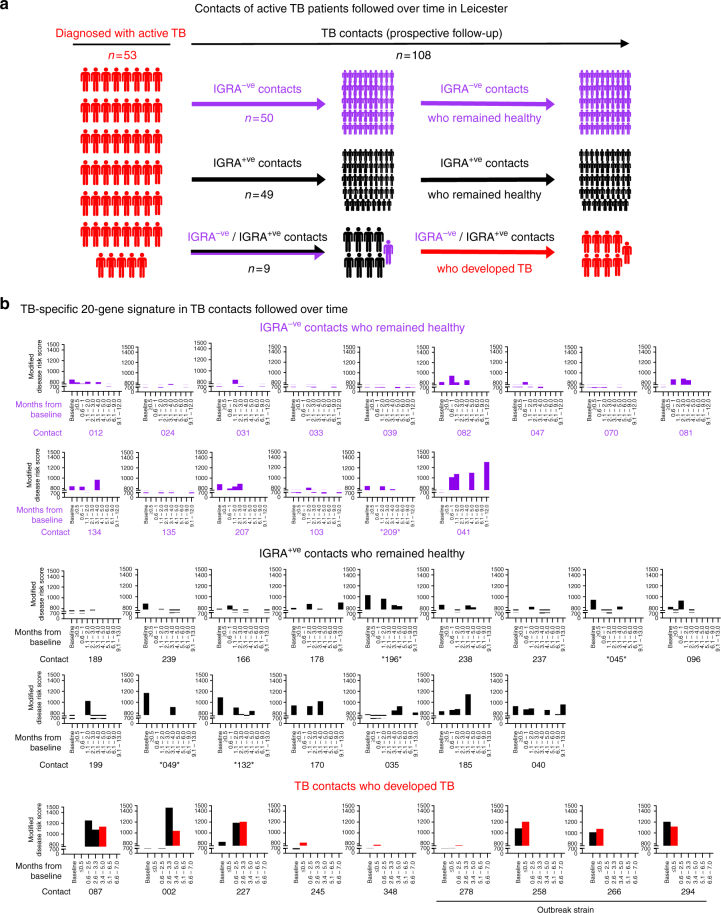
Whole blood transcriptional signatures distinguishing active tuberculosis patients from asymptomatic latently infected individuals exist. Consensus has not been achieved regarding the optimal reduced gene sets as diagnostic biomarkers that also achieve discrimination from other diseases. Here we show a blood transcriptional signature of active tuberculosis using RNA-Seq, confirming microarray results, that discriminates active tuberculosis from latently infected and healthy individuals, validating this signature in an independent cohort. Using an advanced modular approach, we utilise the information from the entire transcriptome, which includes overabundance of type I interferon-inducible genes and underabundance of IFNG and TBX21, to develop a signature that discriminates active tuberculosis patients from latently infected individuals or those with acute viral and bacterial infections. We suggest that methods targeting gene selection across multiple discriminant modules can improve the development of diagnostic biomarkers with improved performance. Finally, utilising the modular approach, we demonstrate dynamic heterogeneity in a longitudinal study of recent tuberculosis contacts.
Conflict of interest statement
The authors declare no competing interests and note that previous patents held by A.O.G. on the use of the blood transcriptomic for diagnosis of tuberculosis have lapsed and discontinued. Neither bioMérieux nor BIOASTER have filed patents related to this study.
Figures
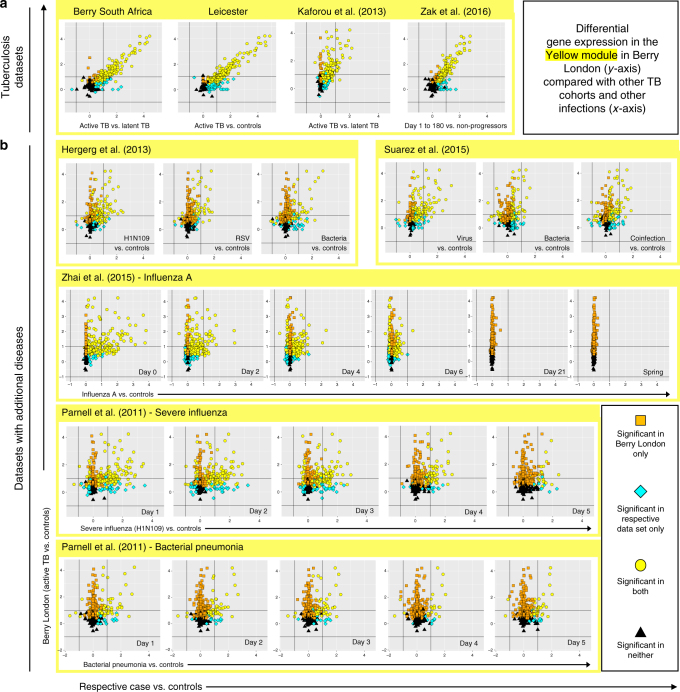
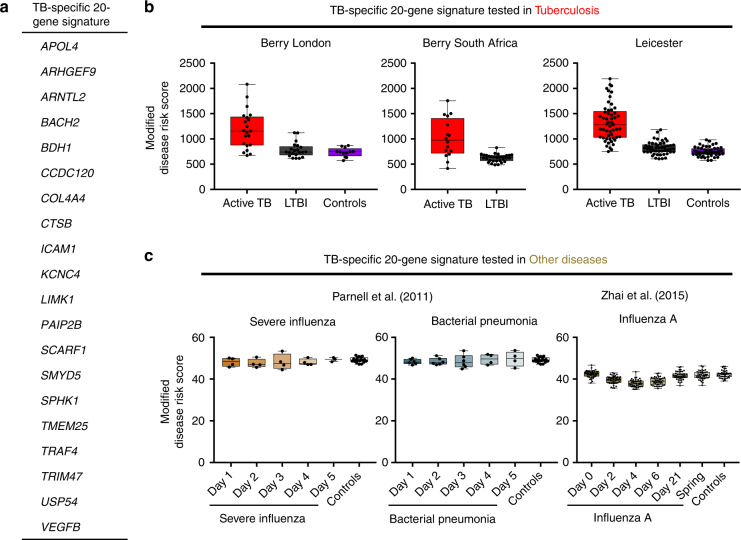
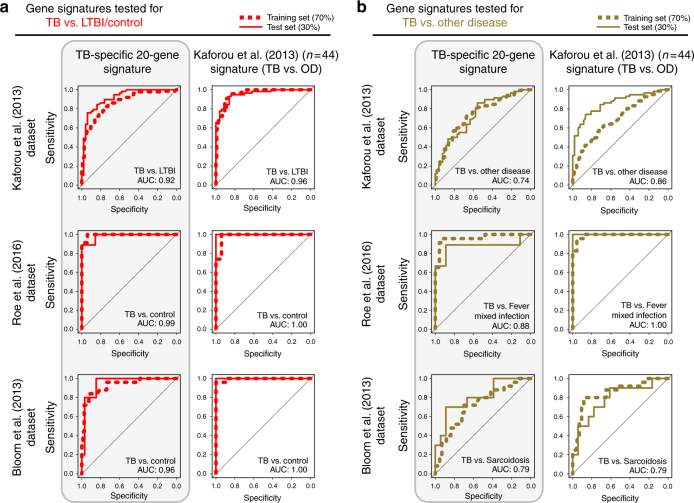
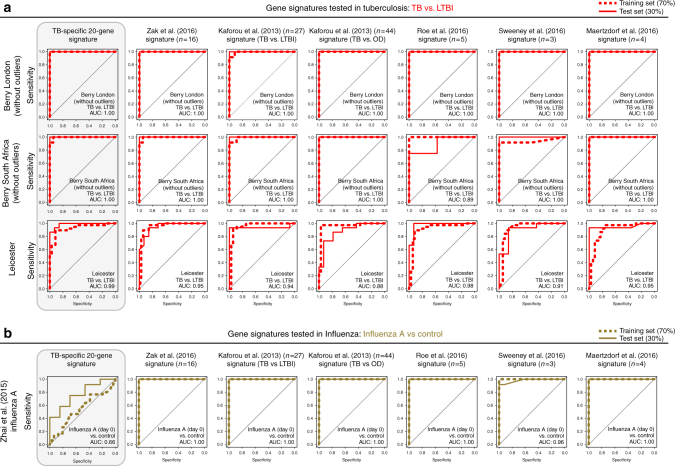

References
-
- World Health Organisation. Global TB Report (WHO, Geneva, 2015).
-
- Center for Communicable Disease Control and Prevention. Reported Tuberculosis in the United States, 2007. (US Department of Health and Human Services, Atlanta, GA, 2007).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
