

Comparative Genomic Analysis of Re-emergent Human Adenovirus Type 55 Pathogens Associated With Adult Severe Community-Acquired Pneumonia Reveals Conserved Genomes and Capsid Proteins
- PMID: 29922263
- PMCID: PMC5996824
- DOI: 10.3389/fmicb.2018.01180
Comparative Genomic Analysis of Re-emergent Human Adenovirus Type 55 Pathogens Associated With Adult Severe Community-Acquired Pneumonia Reveals Conserved Genomes and Capsid Proteins
Abstract
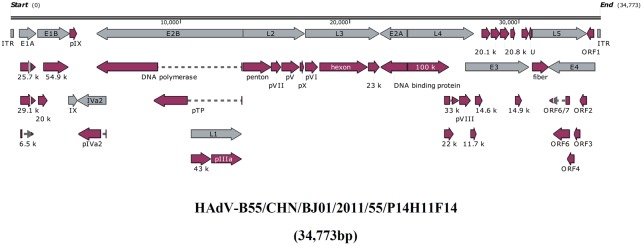
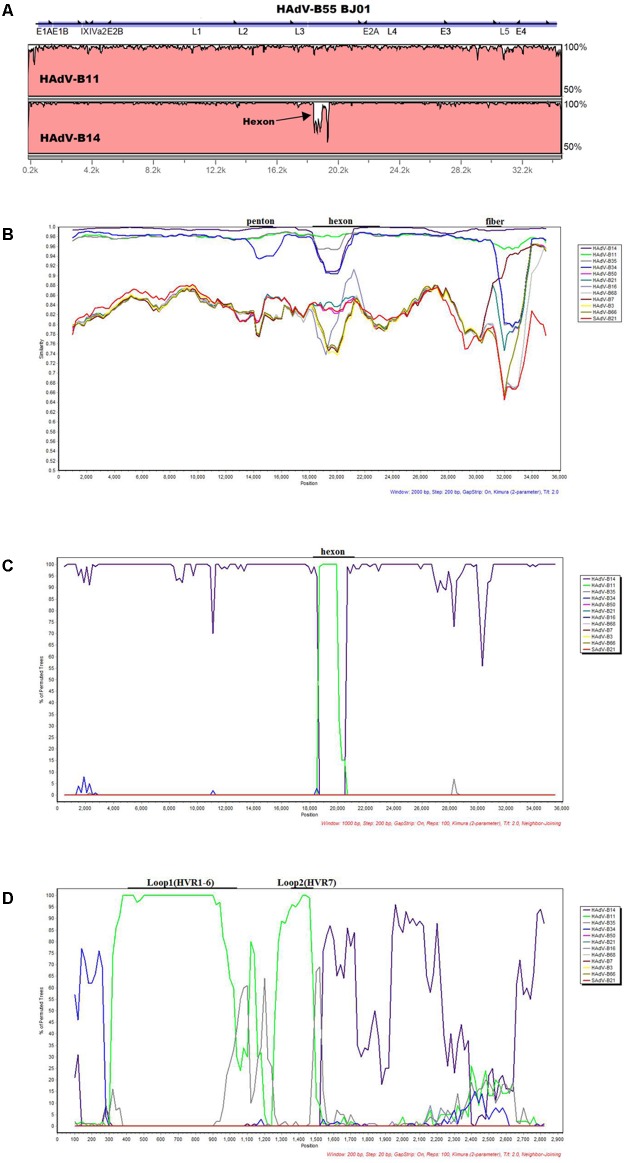
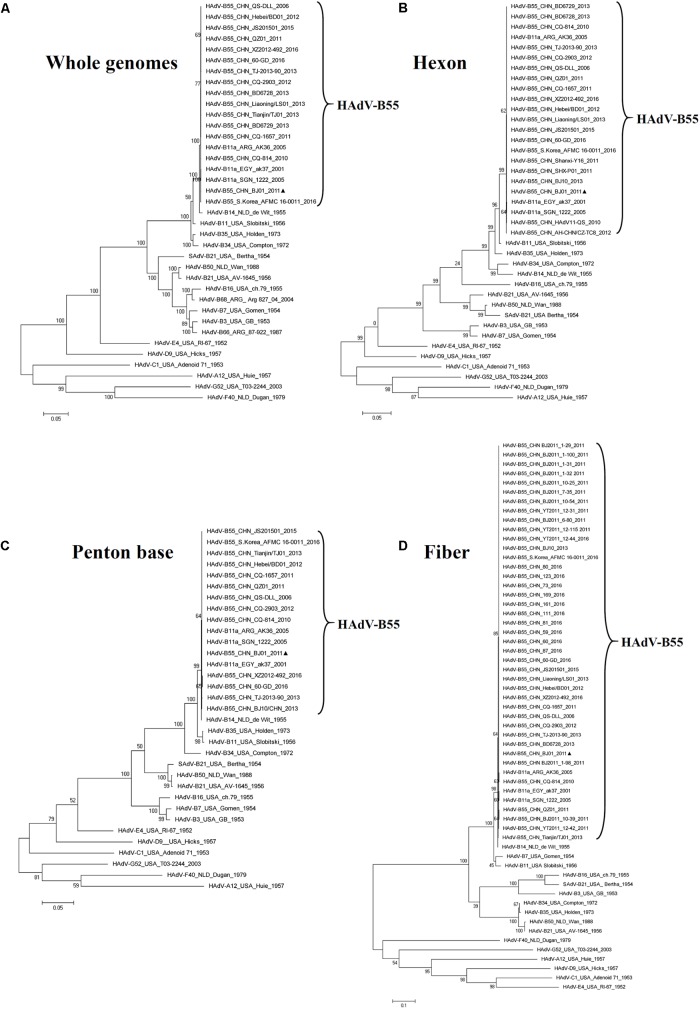
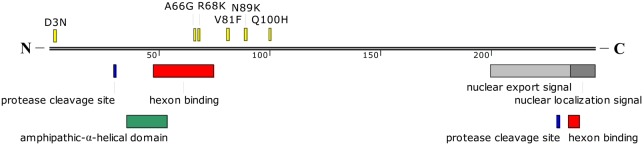
Human adenovirus type 55 (HAdV-B55) is a recently identified acute respiratory disease (ARD) pathogen in HAdV species B with a recombinant genome between renal HAdV-B11 and respiratory HAdV-B14. Since HAdV-B55 first appeared in China school in 2006, no more ARD cases associated with it had been reported until 2011, when there was an outbreak of adult severe community-acquired pneumonia (CAP) in Beijing, China. Reported here is the bioinformatics analysis of the re-emergent HAdV-B55 responsible for this outbreak. Recombination and protein sequence analysis re-confirmed that this isolate (BJ01) was a recombinant virus with the capsid hexon gene from HAdV-B11. The selection pressures for the three capsid proteins, i.e., hexon, penton base, and fiber genes, were all negative, along with very low non-synonymous (dN) and synonymous (dS) substitutions/site (<0.0007). Phylogenetic analyses of the whole genome and the three major capsid genes of HAdV-B55 revealed the close phylogenetic relationship among all HAdV-B55 strains. Comparative genomic analysis of this re-emergent HAdV-B55 strain (BJ01; 2011) with the first HAdV-B55 strain (QS-DLL; 2006) showed the high genome identity (99.87%), including 10 single-nucleotide non-synonymous substitutions, 11 synonymous substitutions, 3 insertions, and one deletion in non-coding regions. The major non-synonymous substitutions (6 of 10) occurred in the protein pVI in its L3 region, which protein has different functions at various stages of an adenovirus infection, and may be associated with the population distribution of HAdV-B55 infection. No non-synonymous substitutions were found in the three major capsid proteins, which proteins are responsible for type-specific neutralizing antibodies. Comparative genomic analysis of the re-emergent HAdV-B55 strains associated with adult severe CAP revealed conserved genome and capsid proteins, providing the foundation for the development of effective vaccines against this pathogen. This study also facilitates the further investigation of HAdV-B55 epidemiology, molecular evolution, patterns of pathogen emergence and re-emergence, and the predication of genome recombination between adenoviruses.
Keywords: China; comparative genomics; human adenovirus type 55; recombination; severe community-acquired pneumonia.
Figures
 ).
).
References
-
- Chen Y., Liu F., Wang C., Zhao M., Deng L., Zhong J., et al. (2016). Molecular identification and epidemiological features of human adenoviruses associated with acute respiratory infections in hospitalized children in Southern China, 2012-2013. PLoS One 11:e0155412. 10.1371/journal.pone.0155412 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous