

Heterocyclic Analogs of Sulforaphane Trigger DNA Damage and Impede DNA Repair in Colon Cancer Cells: Interplay of HATs and HDACs
- PMID: 29924908
- PMCID: PMC6553464
- DOI: 10.1002/mnfr.201800228
Heterocyclic Analogs of Sulforaphane Trigger DNA Damage and Impede DNA Repair in Colon Cancer Cells: Interplay of HATs and HDACs
Abstract
Scope: DNA repair inhibitors have broad clinical applications in tumor types with DNA repair defects, including colorectal cancer (CRC). Structural analogs of the anticancer agent sulforaphane (SFN) were investigated as modifiers of histone deacetylase (HDAC) and histone acetyltransferase (HAT) activity, and for effects on DNA damage/repair pertinent to human CRC.
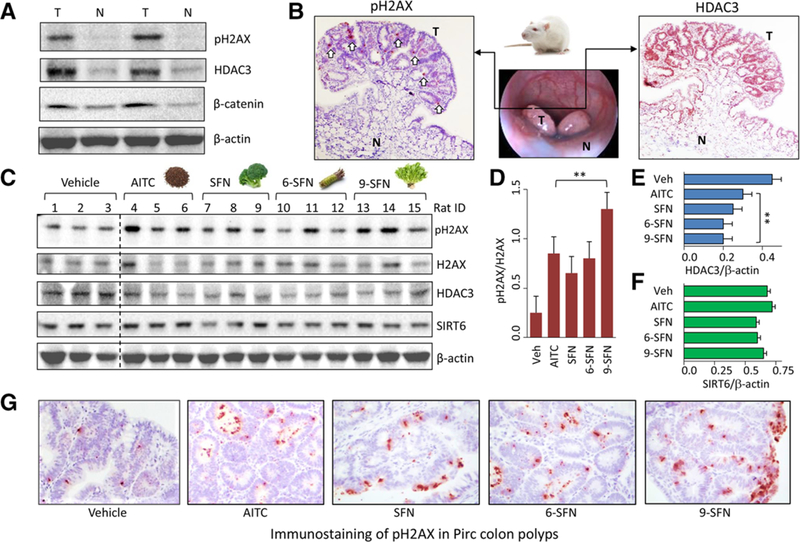
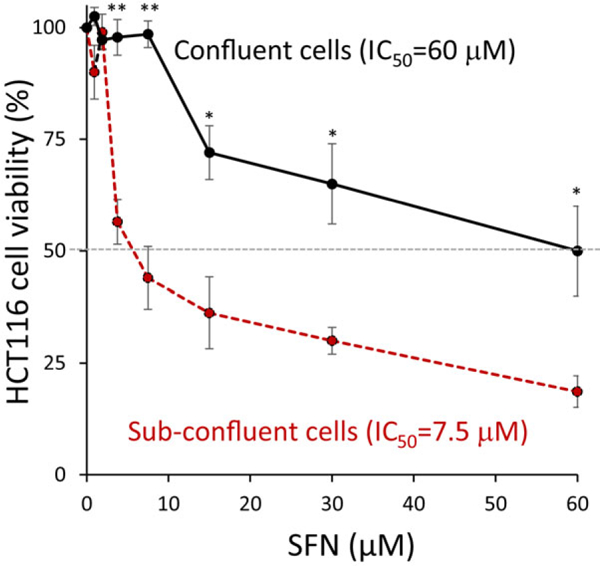
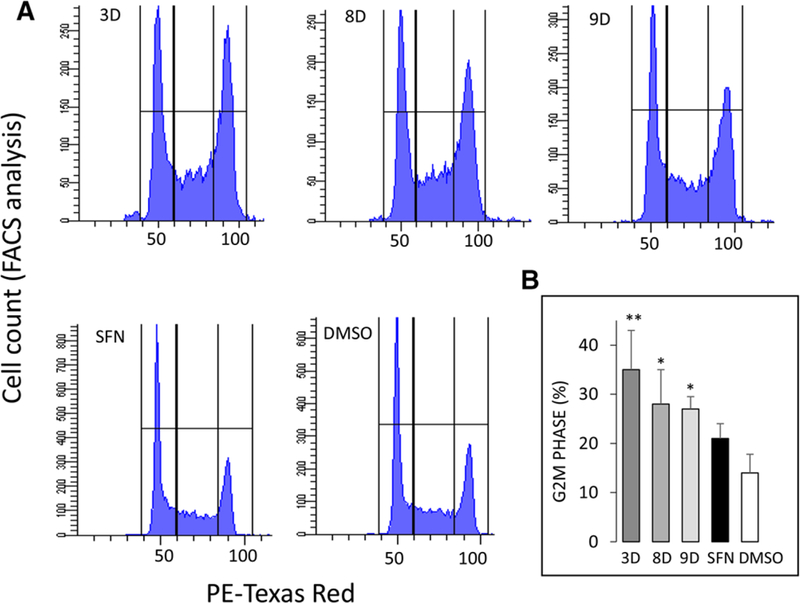
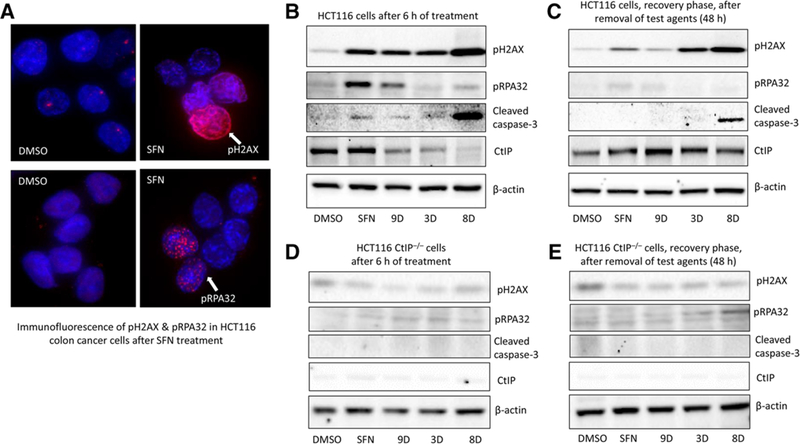
Methods and results: In the polyposis in rat colon (Pirc) model, single oral administration of SFN and structurally related long-chain isothiocyanates (ITCs) decreased histone deacetylase 3 (HDAC3) expression and increased pH2AX levels markedly in adenomatous colon polyps, extending prior observations on HDAC3 inhibition/turnover in cell-based assays. Colon cancer cells at a high initial plating density had diminished cytotoxicity from SFN, whereas novel tetrazole-containing heterocyclic analogs of SFN retained their efficacy. The potent SFN analogs triggered DNA damage, cell cycle arrest, apoptosis, and loss of a key DNA repair regulator, C-terminal binding protein (CtBP) interacting protein (CtIP). These SFN analogs also altered HAT/HDAC activities and histone acetylation status, lowered the expression of HDAC3, P300/CBP-associated factor (PCAF) and lysine acetyltransferase 2A (KAT2A/GCN5), and attenuated homologous recombination (HR)/non-homologous end joining (NHEJ) repair activities in colon cancer cells.
Conclusion: Novel tetrazole-containing heterocyclic analogs of SFN provide a new avenue for chemosensitization in colon cancer cells via modulation of HAT/HDAC activities and associated DNA damage/repair signaling pathways.
Keywords: C-terminal binding protein (CtBP) interacting protein; DNA damage; DNA repair; colon cancer; histone acetyltransferase; histone deacetylase; sulforaphane analogs.
© 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Conflict of interest statement
Conflict of Interest
The authors declare no conflict of interest.
Figures
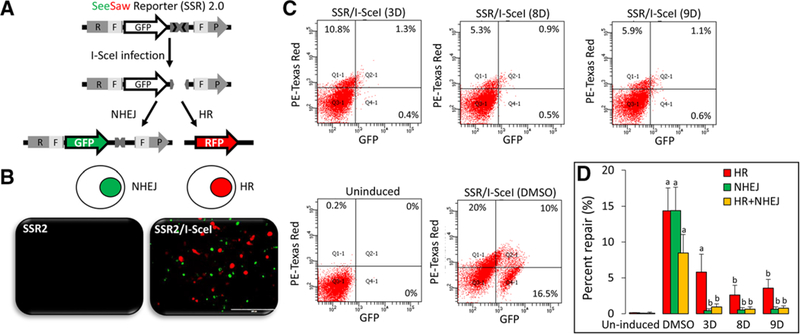
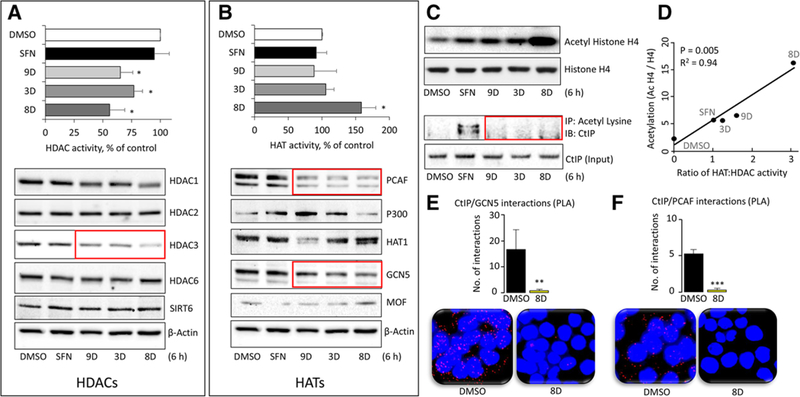
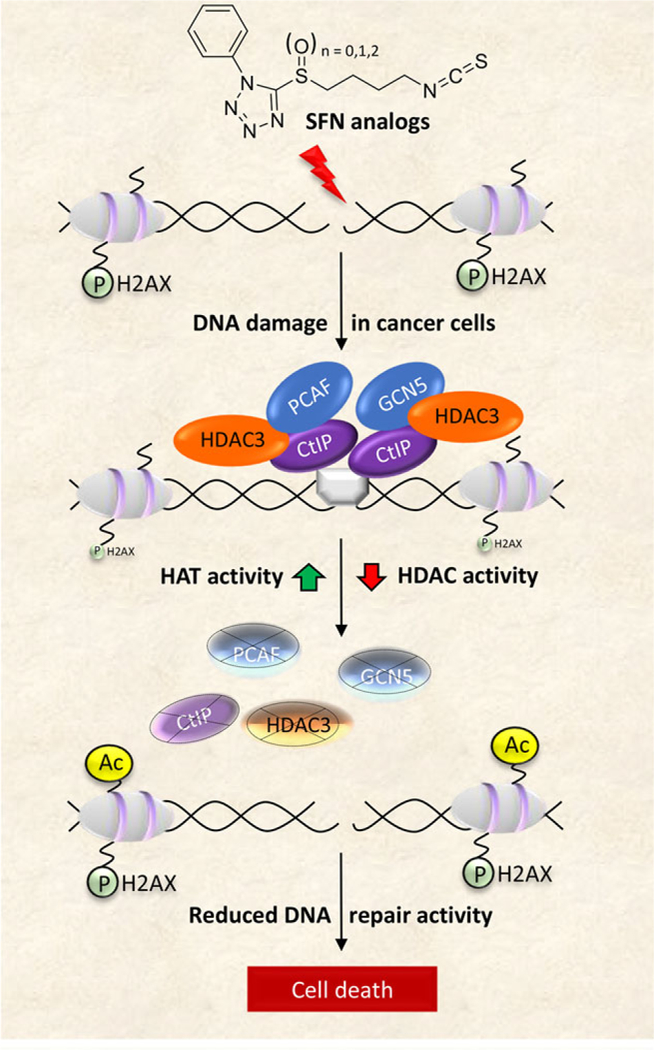
References
-
- American Cancer Society. Cancer Facts & Figures 2018. Atlanta, Ga: American Cancer Society, 2018. Retrieved from https://www.cancer.org/cancer/colon-rectal-cancer/about/key-statistics.html
-
- Lord CJ, Ashworth A, Curr. Opin. Pharmacol. 2008, 8, 363. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
