

ARKS: chromosome-scale scaffolding of human genome drafts with linked read kmers
- PMID: 29925315
- PMCID: PMC6011487
- DOI: 10.1186/s12859-018-2243-x
ARKS: chromosome-scale scaffolding of human genome drafts with linked read kmers
Abstract
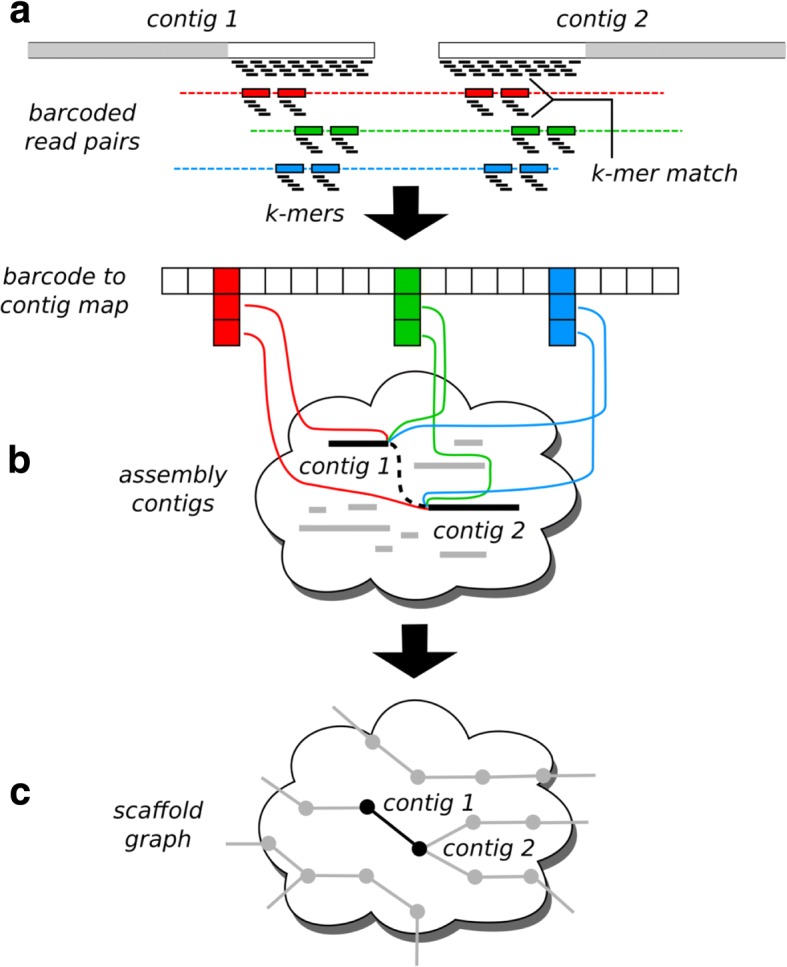
Background: The long-range sequencing information captured by linked reads, such as those available from 10× Genomics (10xG), helps resolve genome sequence repeats, and yields accurate and contiguous draft genome assemblies. We introduce ARKS, an alignment-free linked read genome scaffolding methodology that uses linked reads to organize genome assemblies further into contiguous drafts. Our approach departs from other read alignment-dependent linked read scaffolders, including our own (ARCS), and uses a kmer-based mapping approach. The kmer mapping strategy has several advantages over read alignment methods, including better usability and faster processing, as it precludes the need for input sequence formatting and draft sequence assembly indexing. The reliance on kmers instead of read alignments for pairing sequences relaxes the workflow requirements, and drastically reduces the run time.
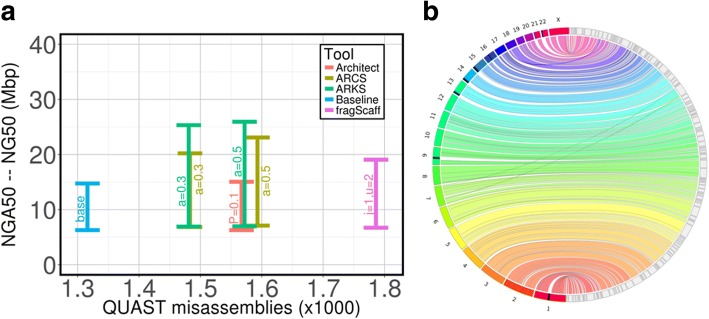
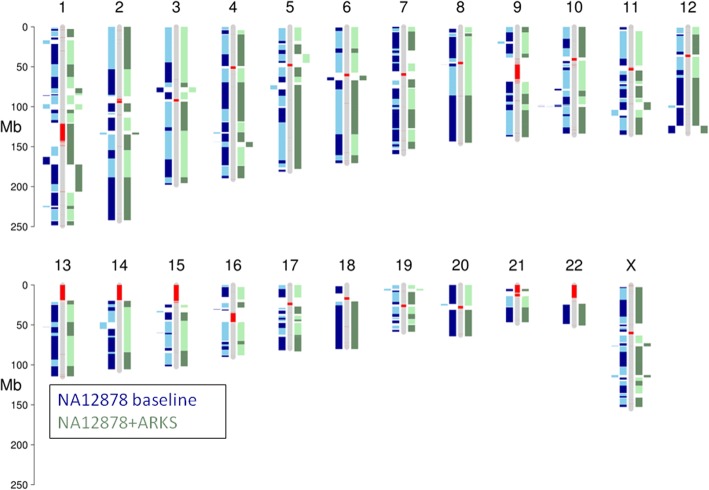
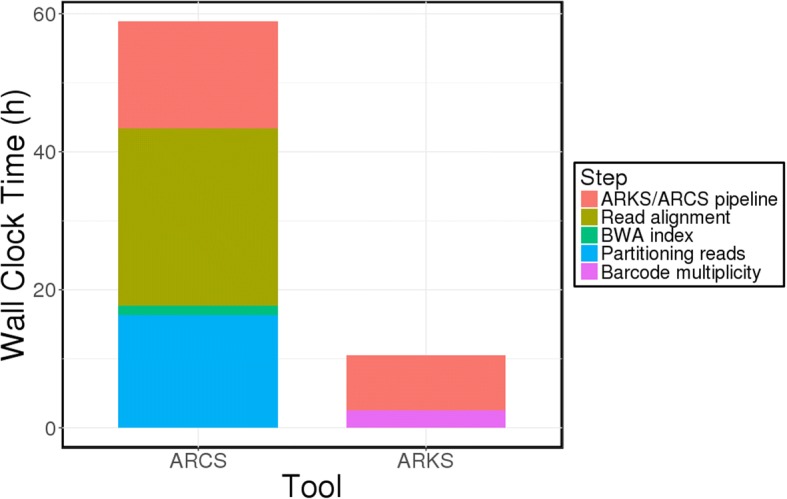
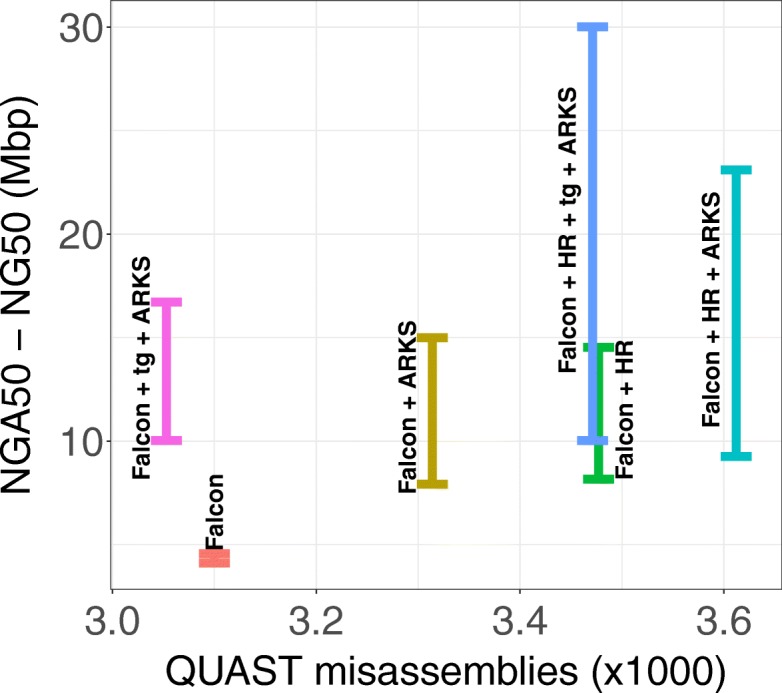
Results: Here, we show how linked reads, when used in conjunction with Hi-C data for scaffolding, improve a draft human genome assembly of PacBio long-read data five-fold (baseline vs. ARKS NG50 = 4.6 vs. 23.1 Mbp, respectively). We also demonstrate how the method provides further improvements of a megabase-scale Supernova human genome assembly (NG50 = 14.74 Mbp vs. 25.94 Mbp before and after ARKS), which itself exclusively uses linked read data for assembly, with an execution speed six to nine times faster than competitive linked read scaffolders (~ 10.5 h compared to 75.7 h, on average). Following ARKS scaffolding of a human genome 10xG Supernova assembly (of cell line NA12878), fewer than 9 scaffolds cover each chromosome, except the largest (chromosome 1, n = 13).
Conclusions: ARKS uses a kmer mapping strategy instead of linked read alignments to record and associate the barcode information needed to order and orient draft assembly sequences. The simplified workflow, when compared to that of our initial implementation, ARCS, markedly improves run time performances on experimental human genome datasets. Furthermore, the novel distance estimator in ARKS utilizes barcoding information from linked reads to estimate gap sizes. It accomplishes this by modeling the relationship between known distances of a region within contigs and calculating associated Jaccard indices. ARKS has the potential to provide correct, chromosome-scale genome assemblies, promptly. We expect ARKS to have broad utility in helping refine draft genomes.
Keywords: 10× Genomics Chromium; ARCS; ARKS; Genome scaffolding; Kmers; Linked reads; Next-generation sequencing; Read mapping; Supernova assembler; de novo assembly.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
ARCS: scaffolding genome drafts with linked reads.Bioinformatics. 2018 Mar 1;34(5):725-731. doi: 10.1093/bioinformatics/btx675. Bioinformatics. 2018. PMID: 29069293 Free PMC article.
-
LongStitch: high-quality genome assembly correction and scaffolding using long reads.BMC Bioinformatics. 2021 Oct 30;22(1):534. doi: 10.1186/s12859-021-04451-7. BMC Bioinformatics. 2021. PMID: 34717540 Free PMC article.
-
Maptcha: an efficient parallel workflow for hybrid genome scaffolding.BMC Bioinformatics. 2024 Aug 8;25(1):263. doi: 10.1186/s12859-024-05878-4. BMC Bioinformatics. 2024. PMID: 39118013 Free PMC article.
-
The Bioinformatic Applications of Hi-C and Linked Reads.Genomics Proteomics Bioinformatics. 2024 Oct 15;22(4):qzae048. doi: 10.1093/gpbjnl/qzae048. Genomics Proteomics Bioinformatics. 2024. PMID: 38905513 Free PMC article. Review.
-
New Approaches for Genome Assembly and Scaffolding.Annu Rev Anim Biosci. 2019 Feb 15;7:17-40. doi: 10.1146/annurev-animal-020518-115344. Epub 2018 Nov 28. Annu Rev Anim Biosci. 2019. PMID: 30485757 Review.
Cited by
-
SWALO: scaffolding with assembly likelihood optimization.Nucleic Acids Res. 2021 Nov 18;49(20):e117. doi: 10.1093/nar/gkab717. Nucleic Acids Res. 2021. PMID: 34417615 Free PMC article.
-
Identifying the causes and consequences of assembly gaps using a multiplatform genome assembly of a bird-of-paradise.Mol Ecol Resour. 2021 Jan;21(1):263-286. doi: 10.1111/1755-0998.13252. Epub 2020 Oct 10. Mol Ecol Resour. 2021. PMID: 32937018 Free PMC article.
-
SpLitteR: diploid genome assembly using TELL-Seq linked-reads and assembly graphs.PeerJ. 2024 Sep 27;12:e18050. doi: 10.7717/peerj.18050. eCollection 2024. PeerJ. 2024. PMID: 39351368 Free PMC article.
-
Fonio millet genome unlocks African orphan crop diversity for agriculture in a changing climate.Nat Commun. 2020 Sep 8;11(1):4488. doi: 10.1038/s41467-020-18329-4. Nat Commun. 2020. PMID: 32901040 Free PMC article.
-
A Highly Contiguous and Annotated Genome Assembly of the Lesser Prairie-Chicken (Tympanuchus pallidicinctus).Genome Biol Evol. 2023 Apr 6;15(4):evad043. doi: 10.1093/gbe/evad043. Genome Biol Evol. 2023. PMID: 36916502 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
