

Genomic variation and biogeography of Antarctic haloarchaea
- PMID: 29925429
- PMCID: PMC6011602
- DOI: 10.1186/s40168-018-0495-3
Genomic variation and biogeography of Antarctic haloarchaea
Abstract
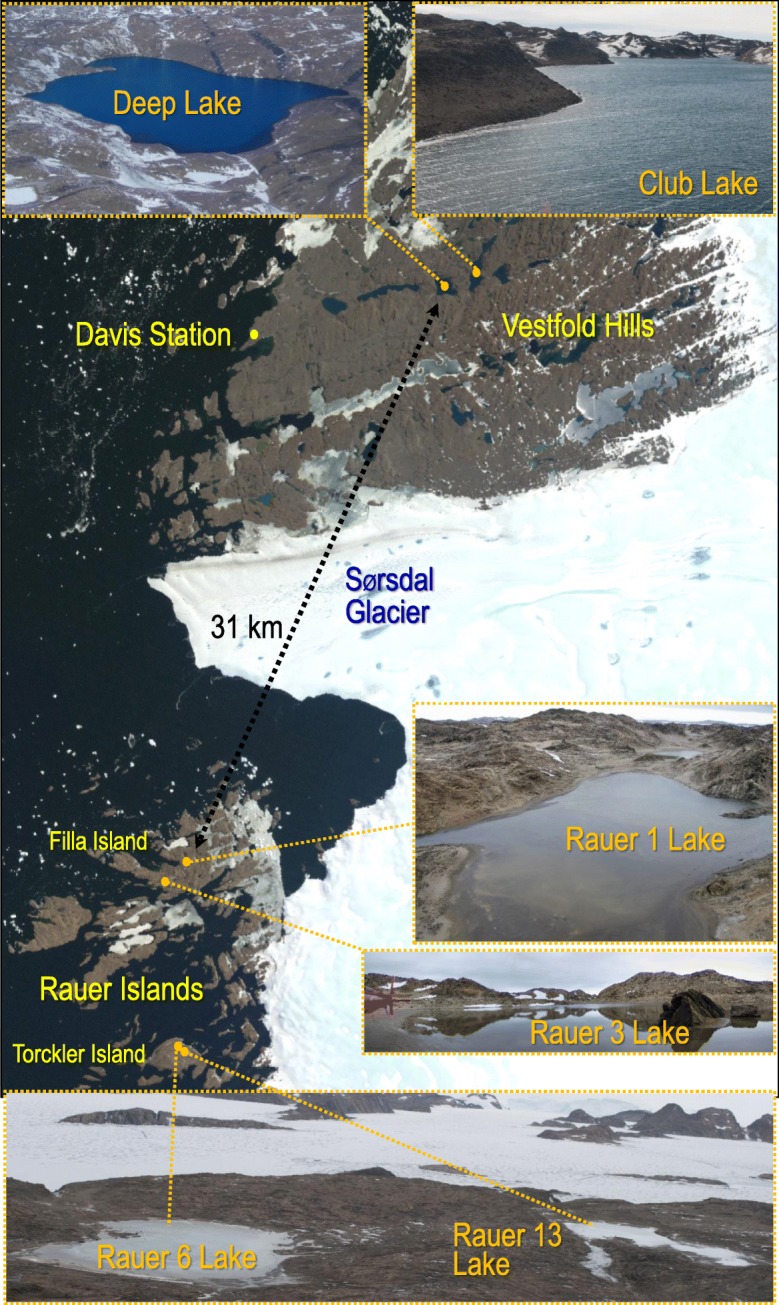
Background: The genomes of halophilic archaea (haloarchaea) often comprise multiple replicons. Genomic variation in haloarchaea has been linked to viral infection pressure and, in the case of Antarctic communities, can be caused by intergenera gene exchange. To expand understanding of genome variation and biogeography of Antarctic haloarchaea, here we assessed genomic variation between two strains of Halorubrum lacusprofundi that were isolated from Antarctic hypersaline lakes from different regions (Vestfold Hills and Rauer Islands). To assess variation in haloarchaeal populations, including the presence of genomic islands, metagenomes from six hypersaline Antarctic lakes were characterised.
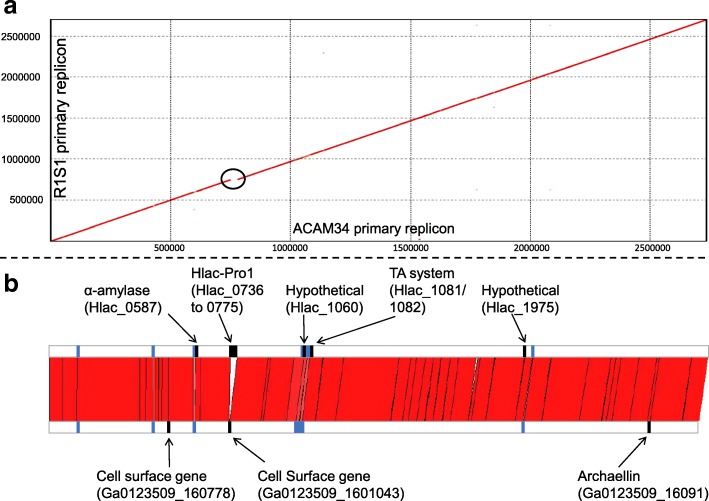
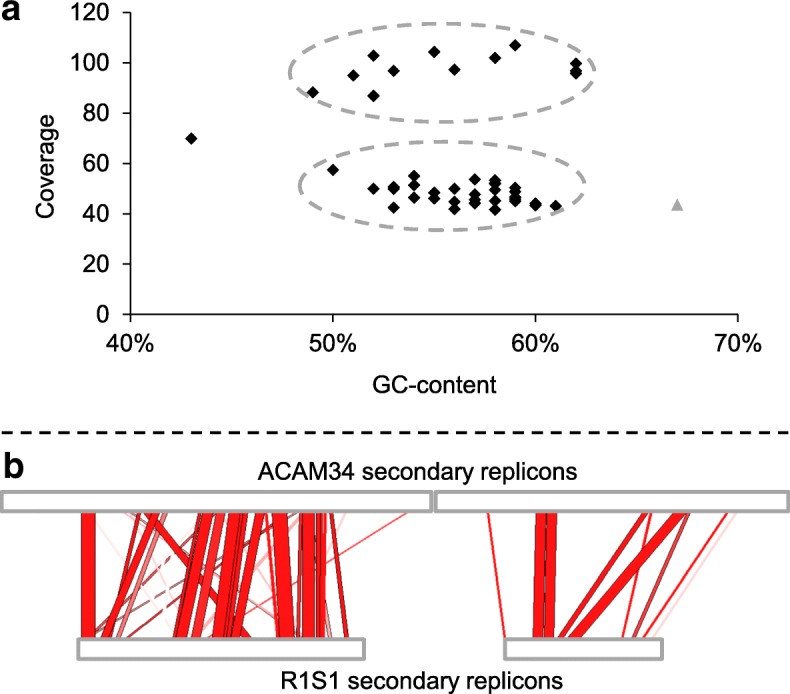
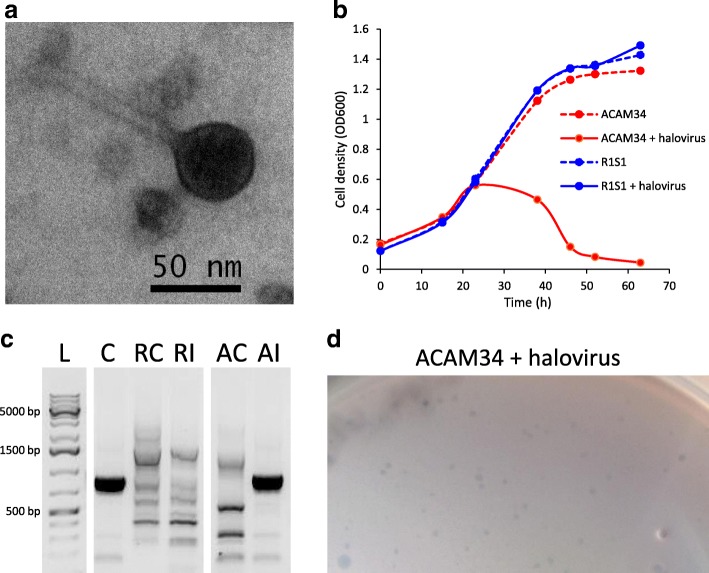
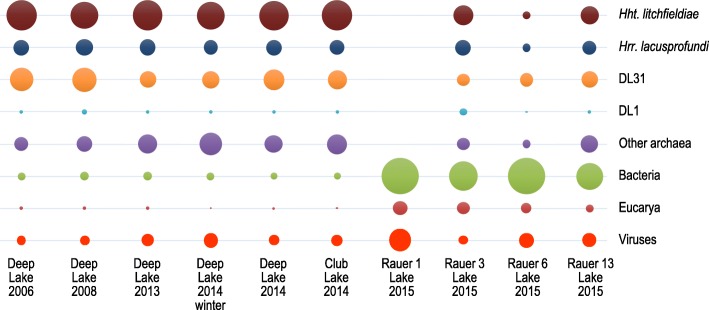
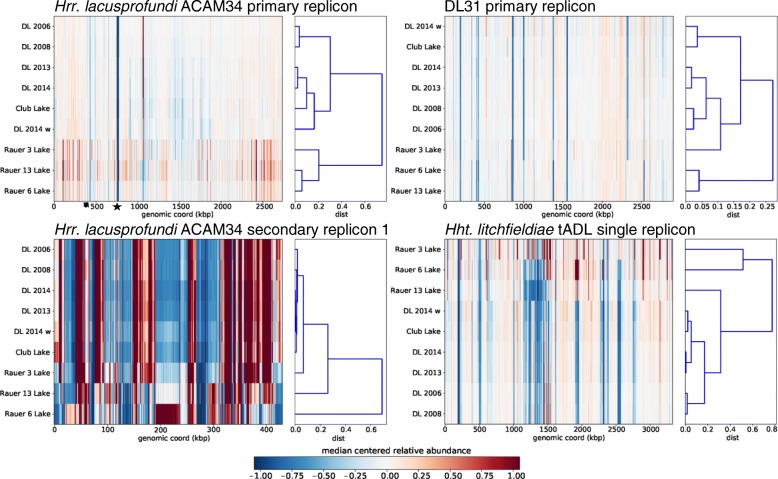
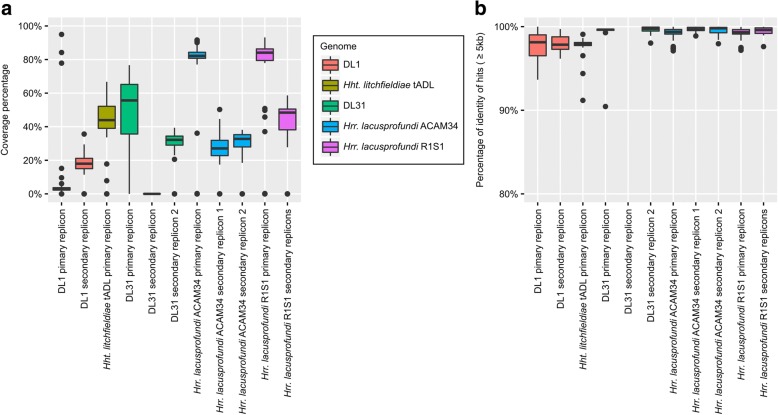
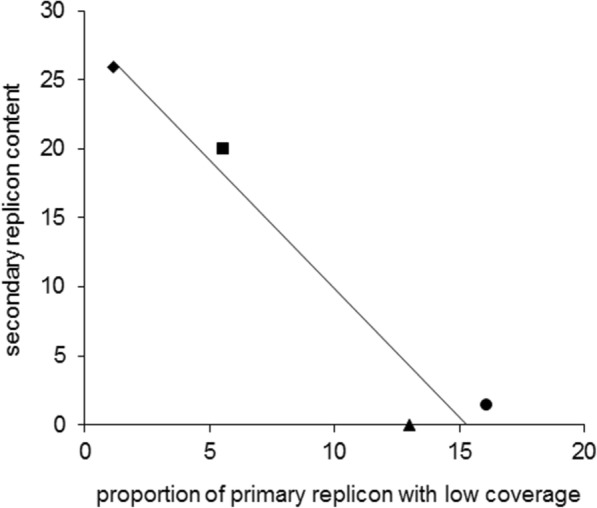
Results: The sequence of the largest replicon of each Hrr. lacusprofundi strain (primary replicon) was highly conserved, while each of the strains' two smaller replicons (secondary replicons) were highly variable. Intergenera gene exchange was identified, including the sharing of a type I-B CRISPR system. Evaluation of infectivity of an Antarctic halovirus provided experimental evidence for the differential susceptibility of the strains, bolstering inferences that strain variation is important for modulating interactions with viruses. A relationship was found between genomic structuring and the location of variation within replicons and genomic islands, demonstrating that the way in which haloarchaea accommodate genomic variability relates to replicon structuring. Metagenome read and contig mapping and clustering and scaling analyses demonstrated biogeographical patterning of variation consistent with environment and distance effects. The metagenome data also demonstrated that specific haloarchaeal species dominated the hypersaline systems indicating they are endemic to Antarctica.
Conclusion: The study describes how genomic variation manifests in Antarctic-lake haloarchaeal communities and provides the basis for future assessments of Antarctic regional and global biogeography of haloarchaea.
Keywords: Antarctica; Biogeography; Genome variation; Genomic islands; Haloarchaea; Halobacteria; Metagenomics; Pan-genome; Replicons; Virus infection.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
