

Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets
- PMID: 29925568
- PMCID: PMC6010767
- DOI: 10.15252/msb.20178124
Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets
Abstract
Multi-omics studies promise the improved characterization of biological processes across molecular layers. However, methods for the unsupervised integration of the resulting heterogeneous data sets are lacking. We present Multi-Omics Factor Analysis (MOFA), a computational method for discovering the principal sources of variation in multi-omics data sets. MOFA infers a set of (hidden) factors that capture biological and technical sources of variability. It disentangles axes of heterogeneity that are shared across multiple modalities and those specific to individual data modalities. The learnt factors enable a variety of downstream analyses, including identification of sample subgroups, data imputation and the detection of outlier samples. We applied MOFA to a cohort of 200 patient samples of chronic lymphocytic leukaemia, profiled for somatic mutations, RNA expression, DNA methylation and ex vivo drug responses. MOFA identified major dimensions of disease heterogeneity, including immunoglobulin heavy-chain variable region status, trisomy of chromosome 12 and previously underappreciated drivers, such as response to oxidative stress. In a second application, we used MOFA to analyse single-cell multi-omics data, identifying coordinated transcriptional and epigenetic changes along cell differentiation.
Keywords: data integration; dimensionality reduction; multi‐omics; personalized medicine; single‐cell omics.
© 2018 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
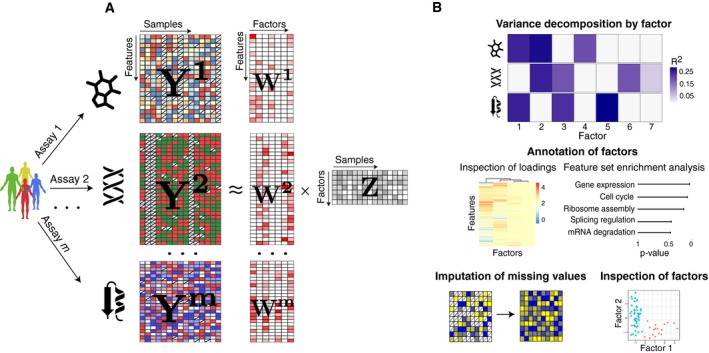
Model overview: MOFA takes M data matrices as input (Y 1,…, Y M), one or more from each data modality, with co‐occurrent samples but features that are not necessarily related and that can differ in numbers. MOFA decomposes these matrices into a matrix of factors (Z) for each sample and M weight matrices, one for each data modality (W 1,.., W M). White cells in the weight matrices correspond to zeros, i.e. inactive features, whereas the cross symbol in the data matrices denotes missing values.
The fitted MOFA model can be queried for different downstream analyses, including (i) variance decomposition, assessing the proportion of variance explained by each factor in each data modality, (ii) semi‐automated factor annotation based on the inspection of loadings and gene set enrichment analysis, (iii) visualization of the samples in the factor space and (iv) imputation of missing values, including missing assays.
- A
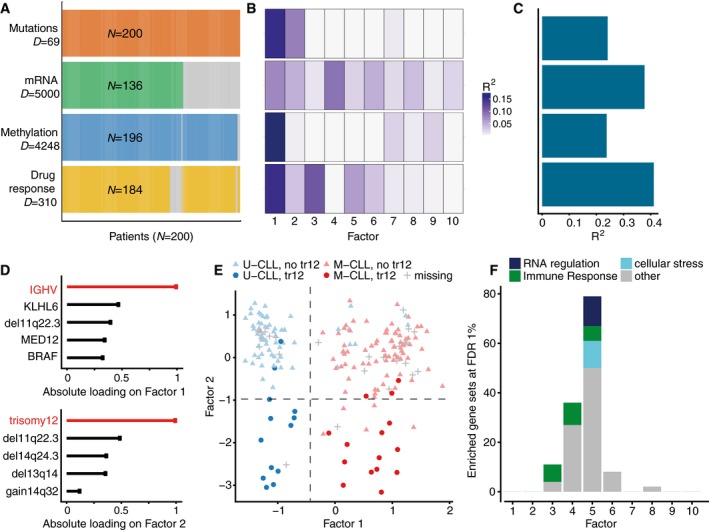
Study overview and data types. Data modalities are shown in different rows (D = number of features) and samples (N) in columns, with missing samples shown using grey bars.
- B, C
(B) Proportion of total variance explained (R 2) by individual factors for each assay and (C) cumulative proportion of total variance explained.
- D
Absolute loadings of the top features of Factors 1 and 2 in the Mutations data.
- E
Visualization of samples using Factors 1 and 2. The colours denote the IGHV status of the tumours; symbol shape and colour tone indicate chromosome 12 trisomy status.
- F
Number of enriched Reactome gene sets per factor based on the gene expression data (FDR < 1%). The colours denote categories of related pathways defined as in Appendix Table S2.
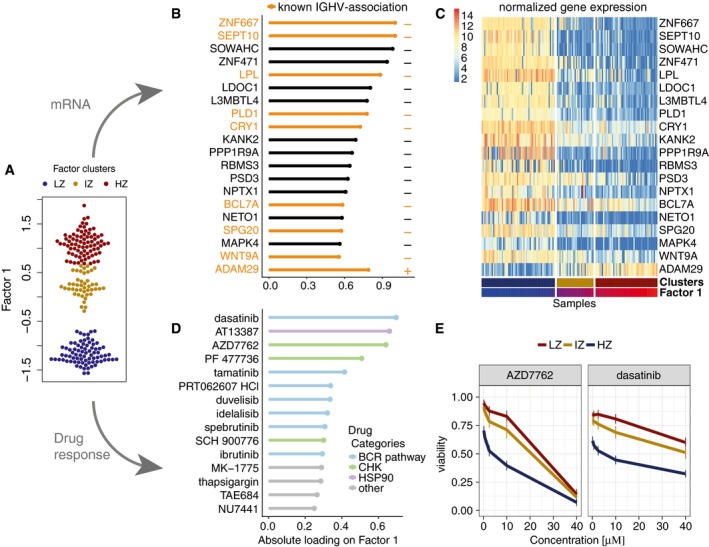
Beeswarm plot with Factor 1 values for each sample with colours corresponding to three groups found by 3‐means clustering with low factor values (LZ), intermediate factor values (IZ) and high factor values (HZ).
Absolute loadings for the genes with the largest absolute weights in the mRNA data. Plus or minus symbols on the right indicate the sign of the loading. Genes highlighted in orange were previously described as prognostic markers in CLL and associated with IGHV status (Vasconcelos et al, 2005; Maloum et al, 2009; Trojani et al, 2012; Morabito et al, 2015; Plesingerova et al, 2017).
Heatmap of gene expression values for genes with the largest weights as in (B).
Absolute loadings of the drugs with the largest weights, annotated by target category.
Drug response curves for two of the drugs with top weights, stratified by the clusters as in (A).
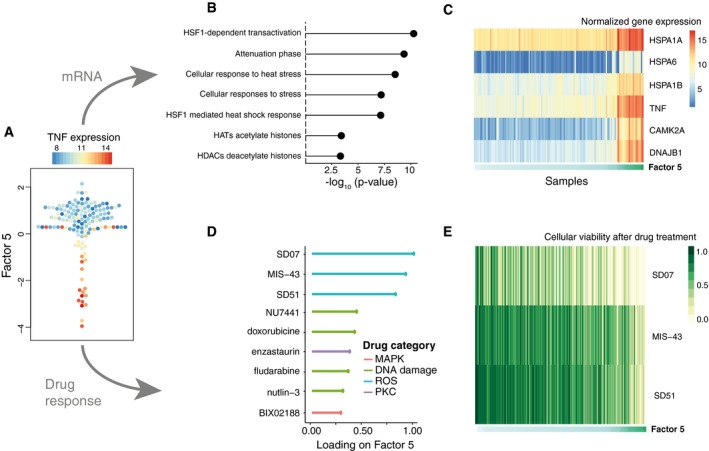
Beeswarm plot of Factor 5. Colours denote the expression of TNF, an inflammatory stress marker.
Gene set enrichment analysis for the top Reactome pathways in the mRNA data (t‐test, Materials and Methods).
Heatmap of gene expression values for the six genes with largest loading. Samples are ordered by their factor values.
Scaled loadings for the top drugs with the largest loading, annotated by target category.
Heatmap of drug response values for the top three drugs with largest loading.
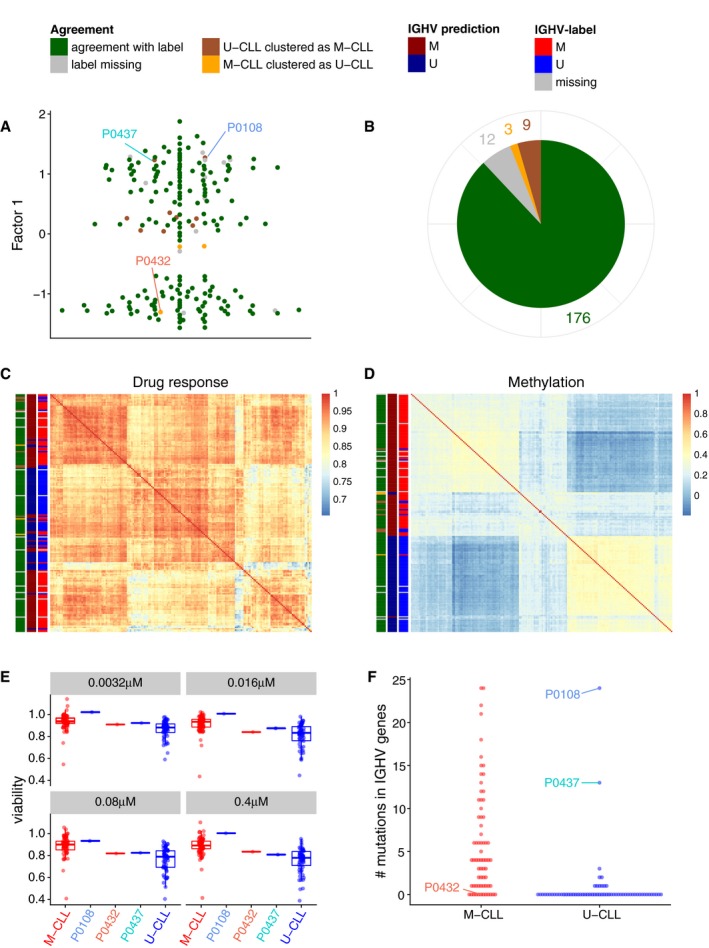
Beeswarm plot of Factor 1 with colours denoting agreement between predicted and clinical labels as in (B).
Pie chart showing total numbers for agreement of imputed labels with clinical label.
Sample‐to‐sample correlation matrix based on drug response data.
Sample‐to‐sample correlation matrix based on methylation data.
Drug response to ONO‐4509 (not included in the training data): Boxplots for the viability values in response to ONO‐4509. The three outlier samples are shown in the middle; on the left and right, the viabilities of the other M‐CLL and U‐CLL samples are shown, respectively. The panels show different drug concentrations tested. Boxes represent the first and third quartiles of the values for M‐CLL and U‐CLL samples, for individual patients the single value.
Whole exome sequencing data on IGHV genes (not included in the training data): the number of mutations found on IGHV genes using whole exome sequencing is shown on the y‐axis, separately for U‐CLL and M‐CLL samples. The three outlier samples are labelled.
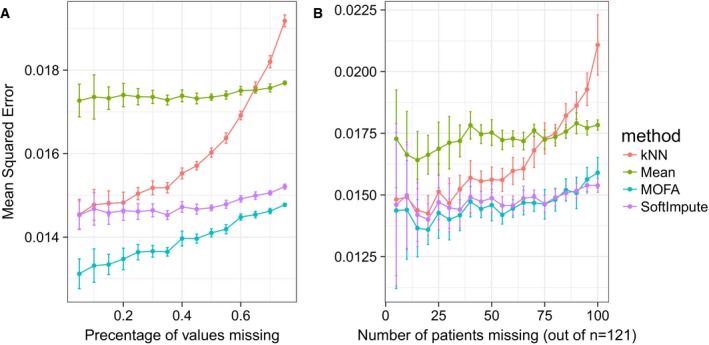
- A, B
Considered were MOFA, SoftImpute, imputation by feature‐wise mean (Mean) and k‐nearest neighbour (kNN). Shown are averages of the mean squared error (MSE) across 15 imputation experiments for increasing fractions of missing data, considering (A) values missing at random and (B) entire assay missing for samples at random. Error bars denote plus or minus two standard error.
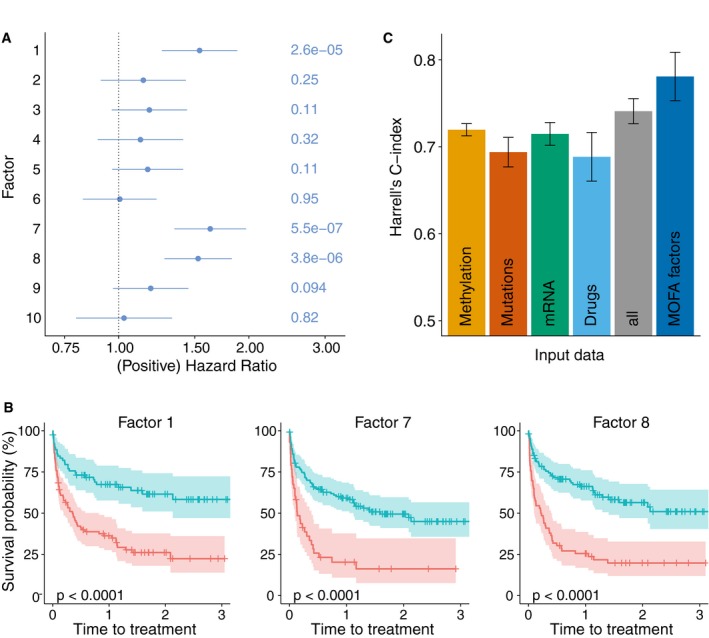
Association of MOFA factors to time to next treatment using a univariate Cox regression with N = 174 samples (96 of which are uncensored cases) and P‐values based on the Wald statistic. Error bars denote 95% confidence intervals. Numbers on the right denote P‐values for each predictor.
Kaplan–Meier plots measuring time to next treatment for the individual MOFA factors. The cut‐points on each factor were chosen using maximally selected rank statistics (Hothorn & Lausen, 2003), and P‐values were calculated using a log‐rank test on the resulting groups.
Prediction accuracy of time to treatment for N = 174 patients using multivariate Cox regression trained using the 10 factors derived using MOFA, as well using the first 10 components obtained from PCA applied to the corresponding single data modalities and the full data set (assessed on hold‐out data). Shown are average values of Harrell's C‐index from fivefold cross‐validation. Error bars denote standard error of the mean.
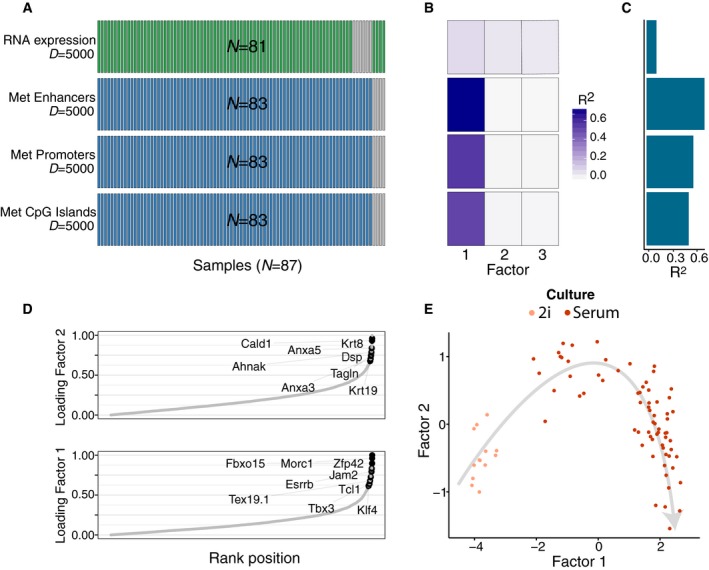
- A
Study overview and data types. Data modalities are shown in different rows (D = number of features) and samples (N) in columns, with missing samples shown using grey bars.
- B, C
(B) Fraction of the variance explained (R 2) by individual factors for each data modality and (C) cumulative proportion of variance explained.
- D
Absolute loadings of Factor 1 (bottom) and Factor 2 (top) in the mRNA data. Labelled genes in Factor 1 are known markers of pluripotency (Mohammed et al, 2017) and genes labelled in Factor 2 are known differentiation markers (Fuchs, 1988).
- E
Scatterplot of Factors 1 and 2. Colours denote culture conditions. The grey arrow illustrates the differentiation trajectory from naive pluripotent cells via primed pluripotent cells to differentiated cells.
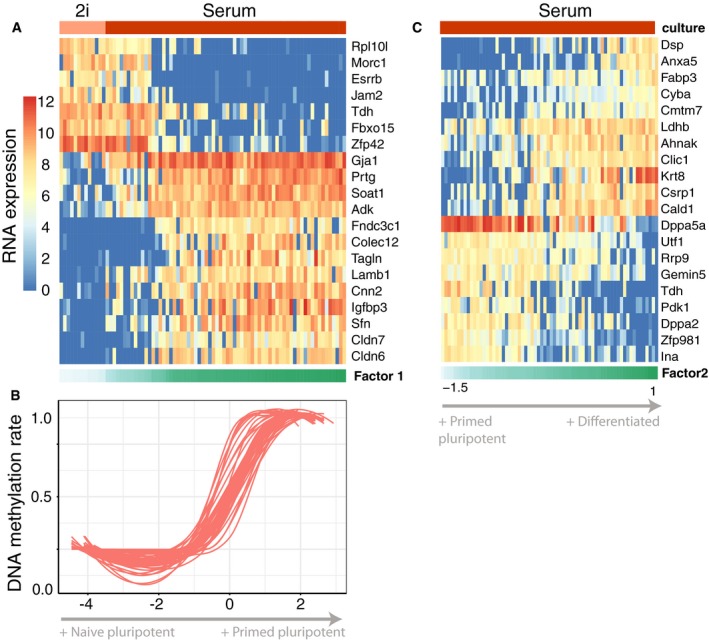
RNA expression changes for the top 20 genes with largest weight on Factor 1.
DNA methylation rate changes for the top 20 CpG sites with largest weight. Shown is a non‐linear loess regression model fit per CpG site.
RNA expression changes for the top 20 genes with largest weight on Factor 2.
References
-
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
