

Indel-correcting DNA barcodes for high-throughput sequencing
- PMID: 29925596
- PMCID: PMC6142223
- DOI: 10.1073/pnas.1802640115
Indel-correcting DNA barcodes for high-throughput sequencing
Abstract
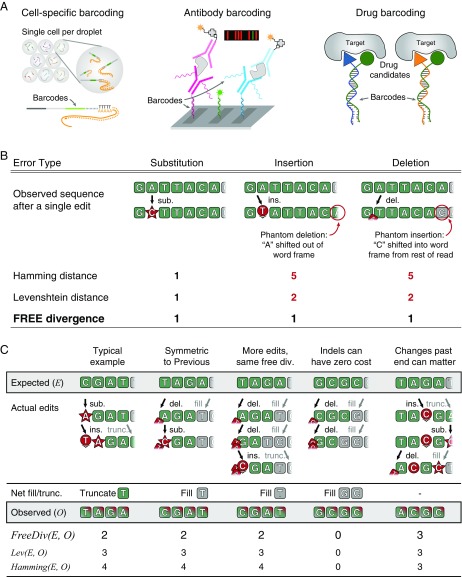
Many large-scale, high-throughput experiments use DNA barcodes, short DNA sequences prepended to DNA libraries, for identification of individuals in pooled biomolecule populations. However, DNA synthesis and sequencing errors confound the correct interpretation of observed barcodes and can lead to significant data loss or spurious results. Widely used error-correcting codes borrowed from computer science (e.g., Hamming, Levenshtein codes) do not properly account for insertions and deletions (indels) in DNA barcodes, even though deletions are the most common type of synthesis error. Here, we present and experimentally validate filled/truncated right end edit (FREE) barcodes, which correct substitution, insertion, and deletion errors, even when these errors alter the barcode length. FREE barcodes are designed with experimental considerations in mind, including balanced guanine-cytosine (GC) content, minimal homopolymer runs, and reduced internal hairpin propensity. We generate and include lists of barcodes with different lengths and error correction levels that may be useful in diverse high-throughput applications, including >106 single-error-correcting 16-mers that strike a balance between decoding accuracy, barcode length, and library size. Moreover, concatenating two or more FREE codes into a single barcode increases the available barcode space combinatorially, generating lists with >1015 error-correcting barcodes. The included software for creating barcode libraries and decoding sequenced barcodes is efficient and designed to be user-friendly for the general biology community.
Keywords: DNA barcodes; error-correcting codes; information storage; massively parallel synthesis.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Kitzman JO. Haplotypes drop by drop. Nat Biotechnol. 2016;34:296–298. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
