

Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity
- PMID: 29925878
- PMCID: PMC6010471
- DOI: 10.1038/s41467-018-04724-5
Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity
Abstract
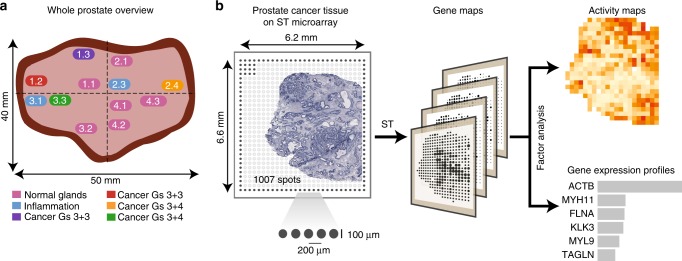
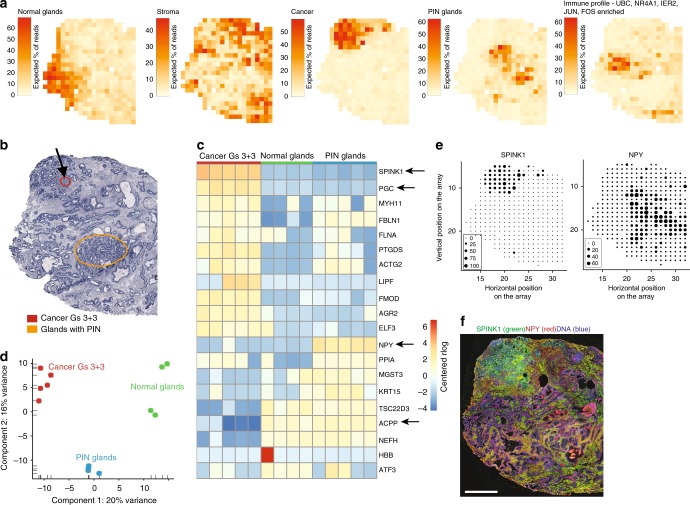
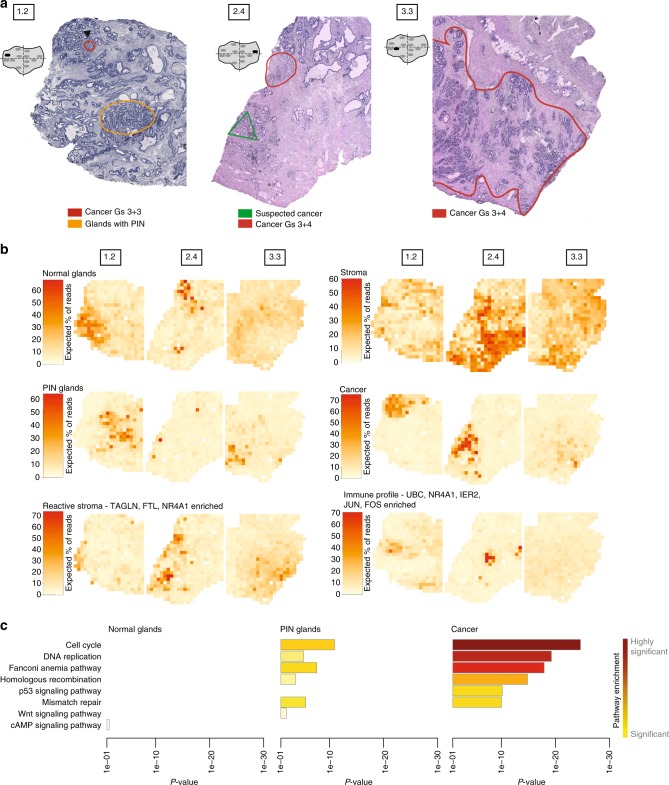
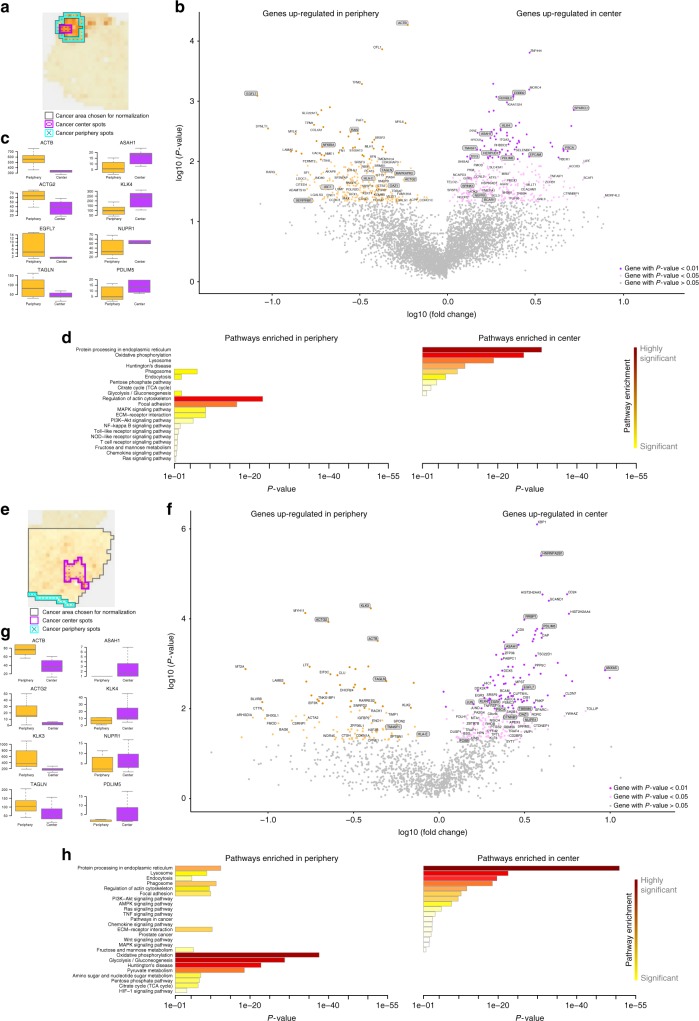
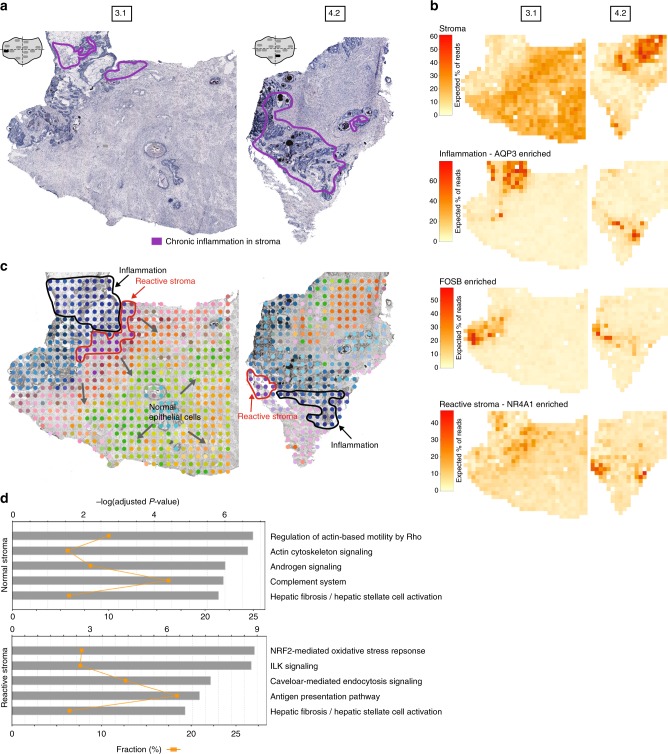
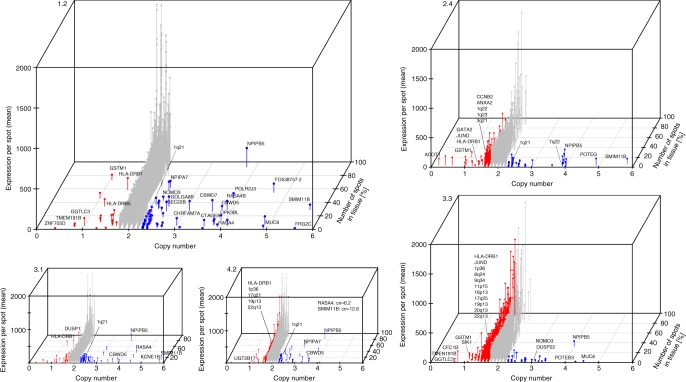
Intra-tumor heterogeneity is one of the biggest challenges in cancer treatment today. Here we investigate tissue-wide gene expression heterogeneity throughout a multifocal prostate cancer using the spatial transcriptomics (ST) technology. Utilizing a novel approach for deconvolution, we analyze the transcriptomes of nearly 6750 tissue regions and extract distinct expression profiles for the different tissue components, such as stroma, normal and PIN glands, immune cells and cancer. We distinguish healthy and diseased areas and thereby provide insight into gene expression changes during the progression of prostate cancer. Compared to pathologist annotations, we delineate the extent of cancer foci more accurately, interestingly without link to histological changes. We identify gene expression gradients in stroma adjacent to tumor regions that allow for re-stratification of the tumor microenvironment. The establishment of these profiles is the first step towards an unbiased view of prostate cancer and can serve as a dictionary for future studies.
Conflict of interest statement
P.L.S., F.S., and J.L. are authors on patents applied for by Spatial Transcriptomics AB covering the technology. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
