

Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation
- PMID: 29925950
- PMCID: PMC6312107
- DOI: 10.1038/s41586-018-0239-3
Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation
Abstract
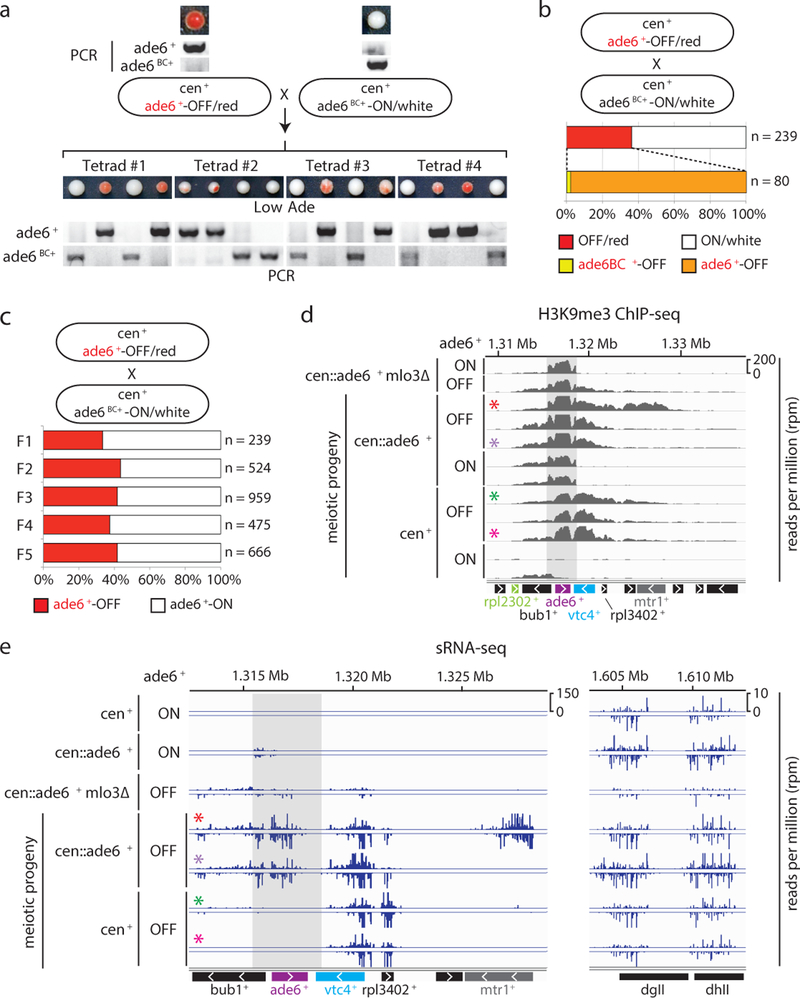
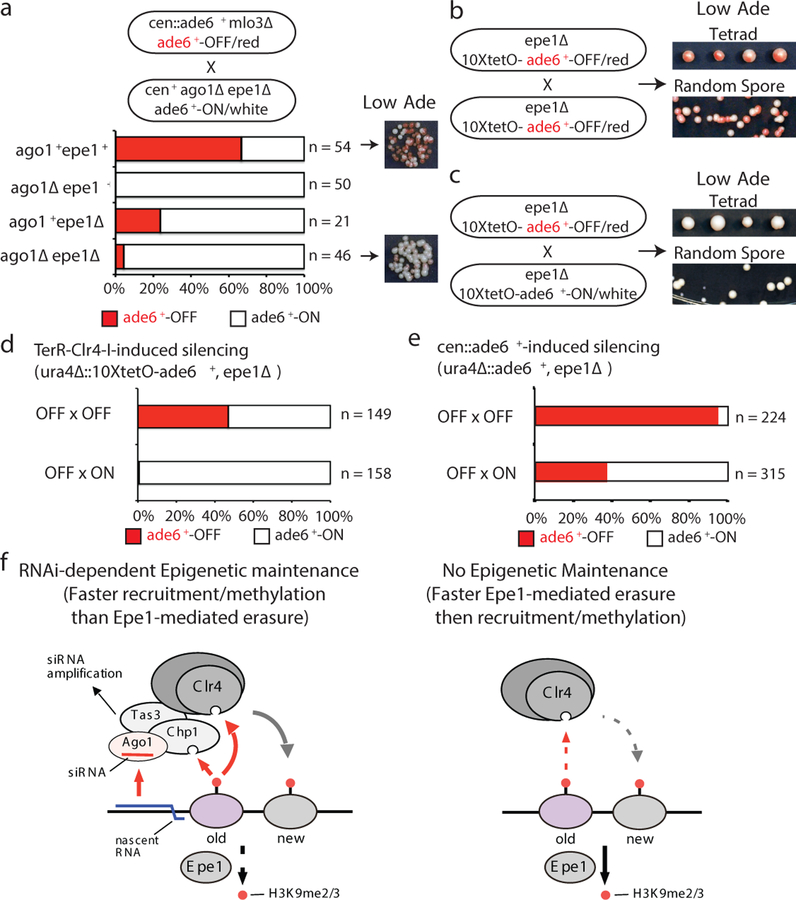
Histone post-translational modifications (PTMs) are associated with epigenetic states that form the basis for cell-type-specific gene expression1,2. Once established, histone PTMs can be maintained by positive feedback involving enzymes that recognize a pre-existing histone modification and catalyse the same modification on newly deposited histones. Recent studies suggest that in wild-type cells, histone PTM-based positive feedback is too weak to mediate epigenetic inheritance in the absence of other inputs3-7. RNA interference (RNAi)-mediated histone H3 lysine 9 methylation (H3K9me) and heterochromatin formation define a potential epigenetic inheritance mechanism in which positive feedback involving short interfering RNA (siRNA) amplification can be directly coupled to histone PTM positive feedback8-14. However, it is not known whether the coupling of these two feedback loops can maintain epigenetic silencing independently of DNA sequence and in the absence of enabling mutations that disrupt genome-wide chromatin structure or transcription15-17. Here, using the fission yeast Schizosaccharomyces pombe, we show that siRNA-induced H3K9me and silencing of a euchromatic gene can be epigenetically inherited in cis during multiple mitotic and meiotic cell divisions in wild-type cells. This inheritance involves the spreading of secondary siRNAs and H3K9me3 to the targeted gene and surrounding areas, and requires both RNAi and H3K9me, suggesting that the siRNA and H3K9me positive-feedback loops act synergistically to maintain silencing. By contrast, when maintained solely by histone PTM positive feedback, silencing is erased by H3K9 demethylation promoted by Epe1, or by interallelic interactions that occur after mating to cells containing an expressed allele even in the absence of Epe1. These findings demonstrate that the RNAi machinery can mediate transgenerational epigenetic inheritance independently of DNA sequence or enabling mutations, and reveal a role for the coupling of the siRNA and H3K9me positive-feedback loops in the protection of epigenetic alleles from erasure.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
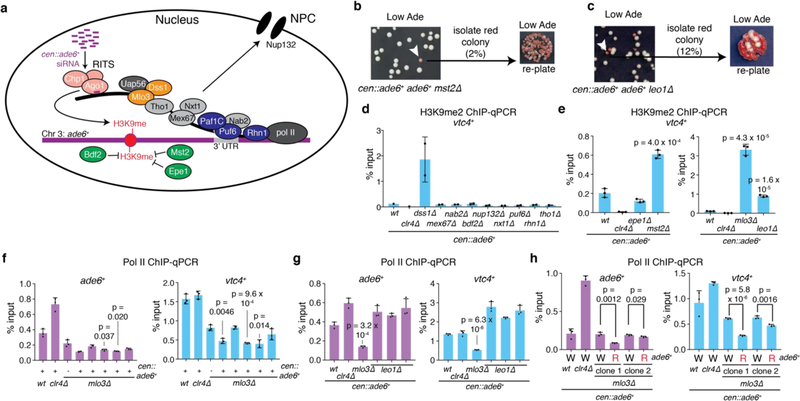
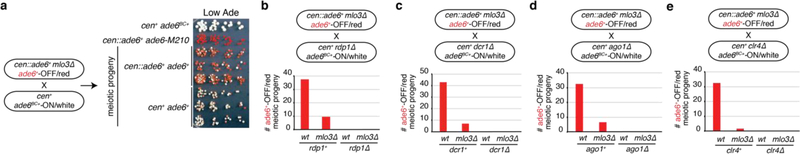
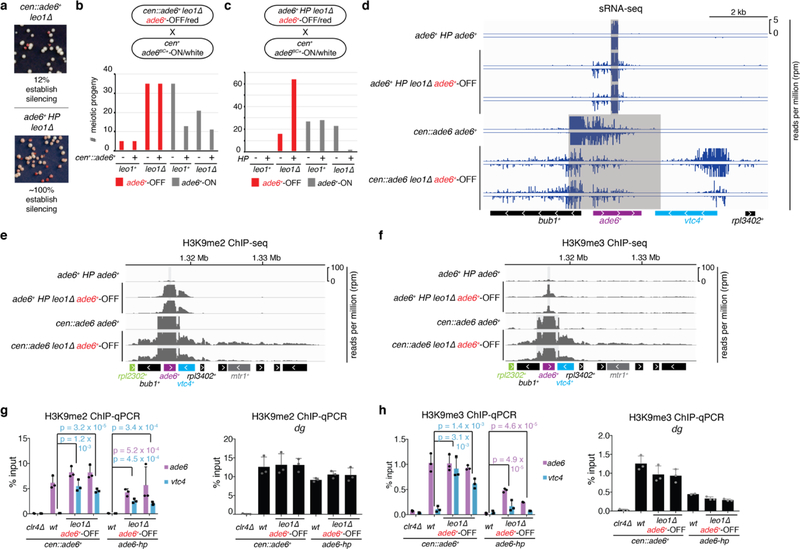
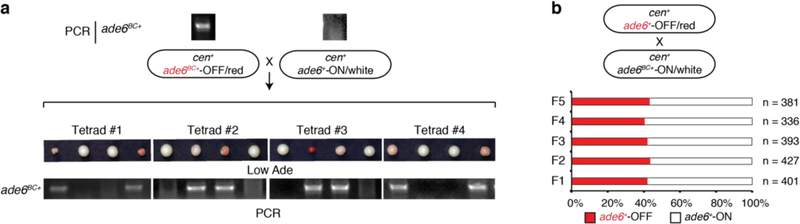
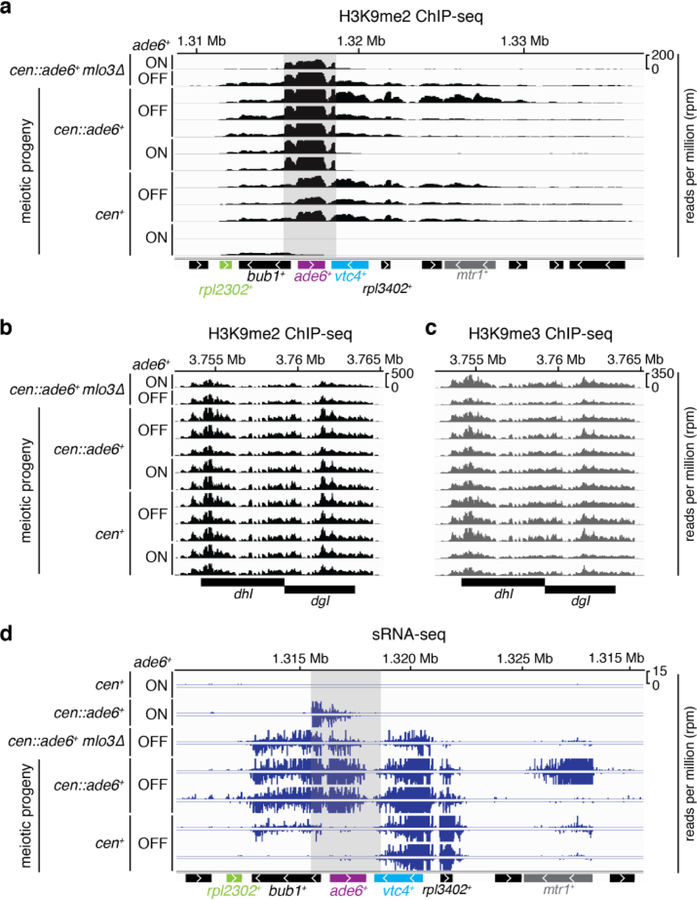
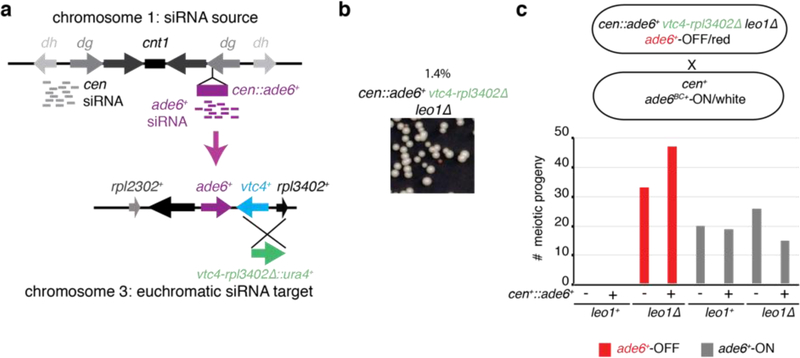
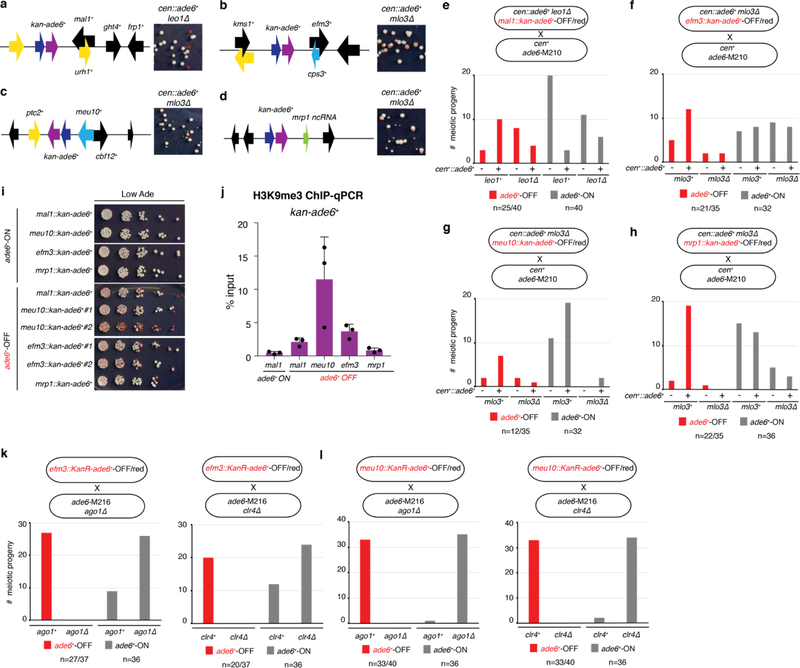
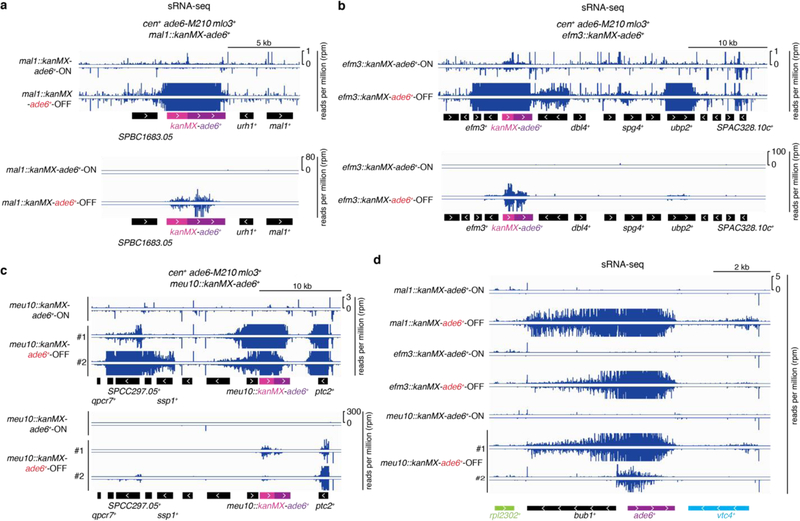
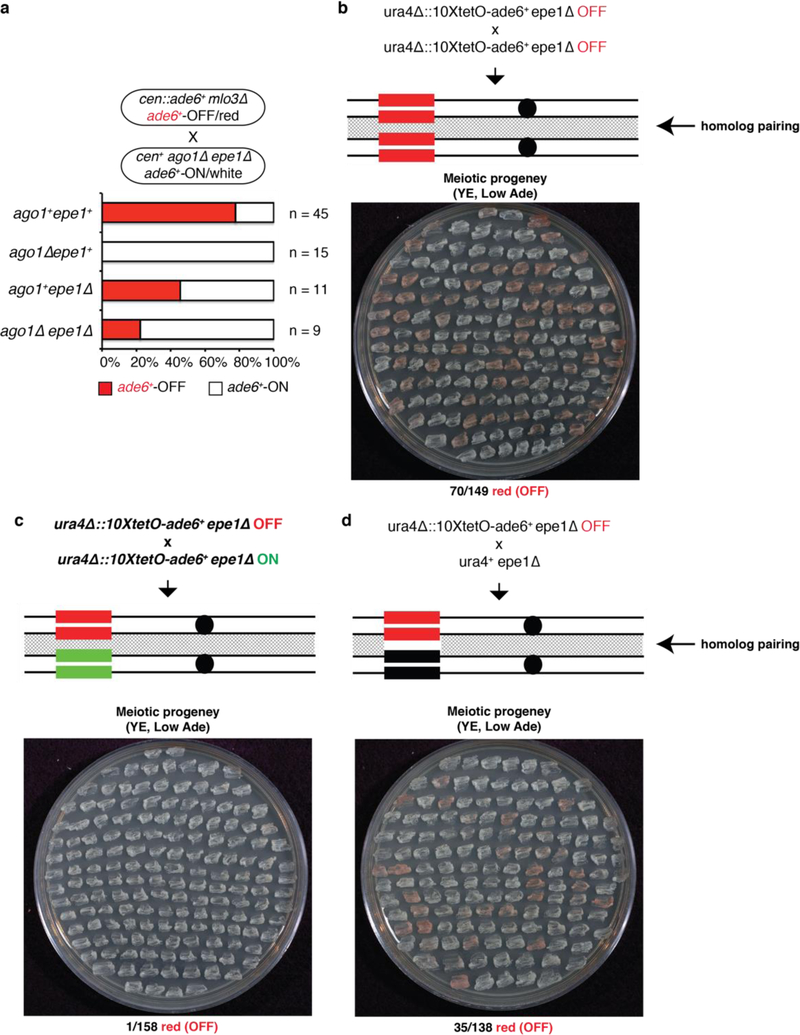
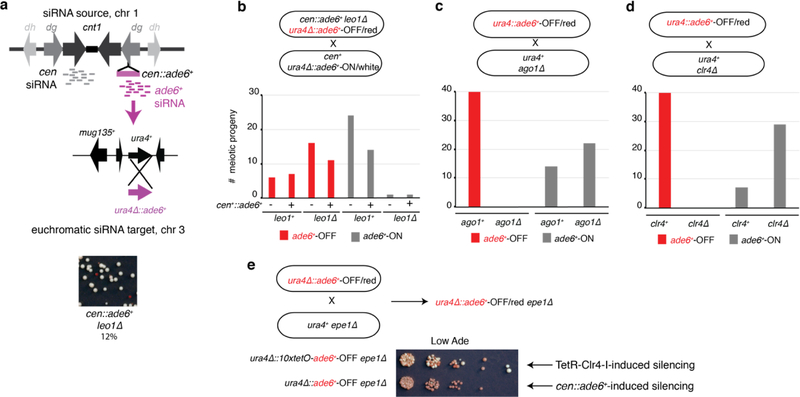
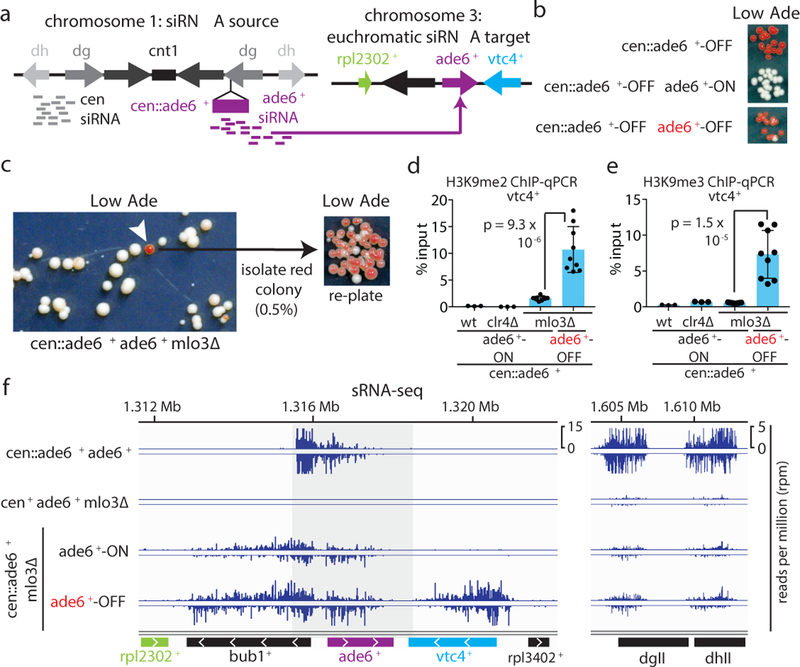
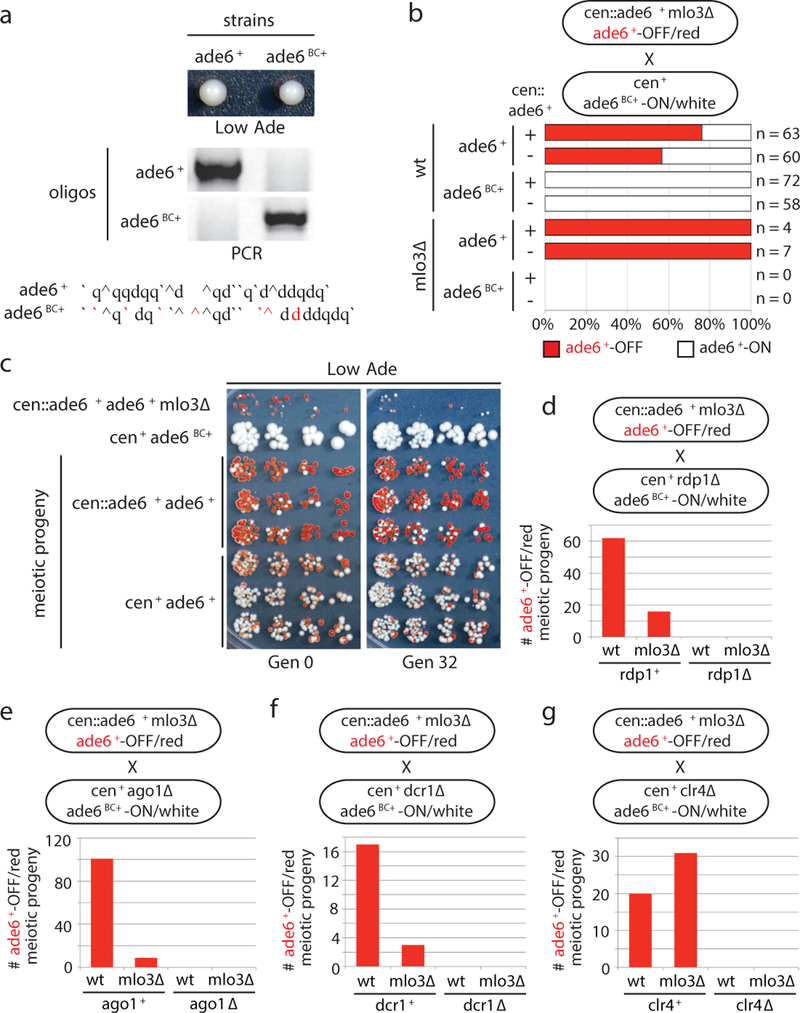
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
