

Improving Prediction Accuracy of Binding Free Energies and Poses of HIV Integrase Complexes Using the Binding Energy Distribution Analysis Method with Flattening Potentials
- PMID: 29927237
- PMCID: PMC6287956
- DOI: 10.1021/acs.jcim.8b00194
Improving Prediction Accuracy of Binding Free Energies and Poses of HIV Integrase Complexes Using the Binding Energy Distribution Analysis Method with Flattening Potentials
Abstract
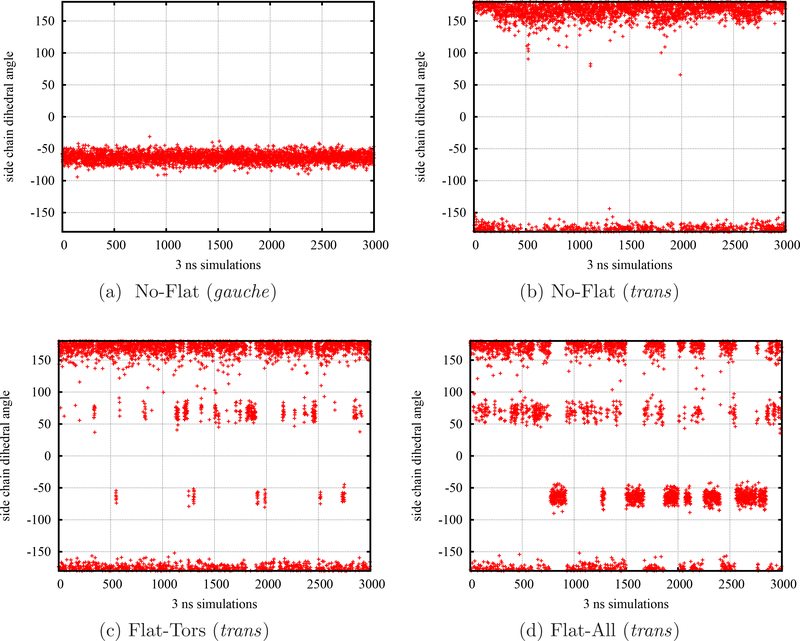
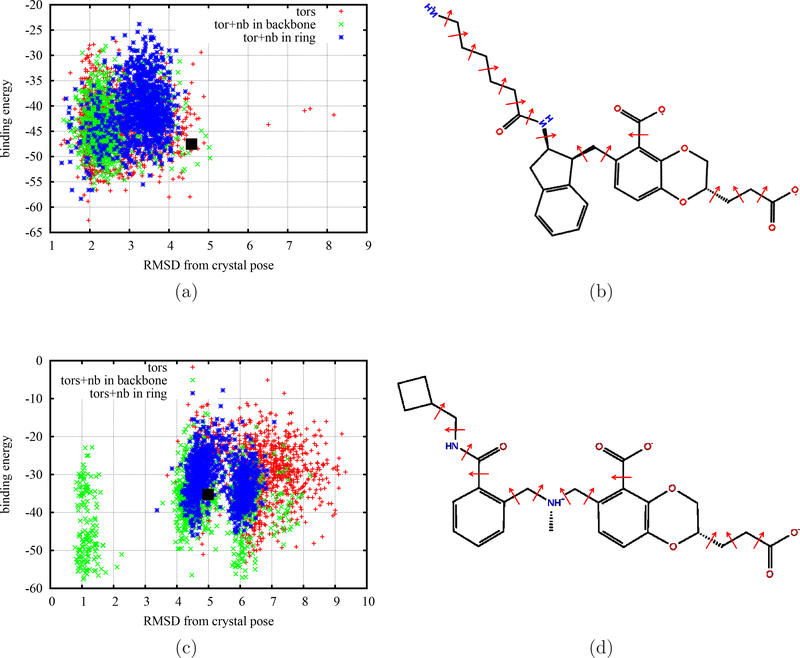
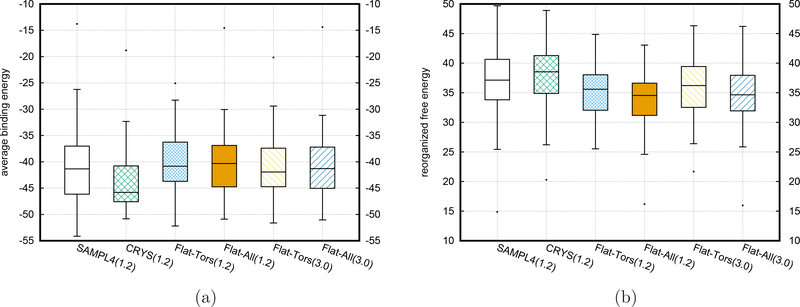
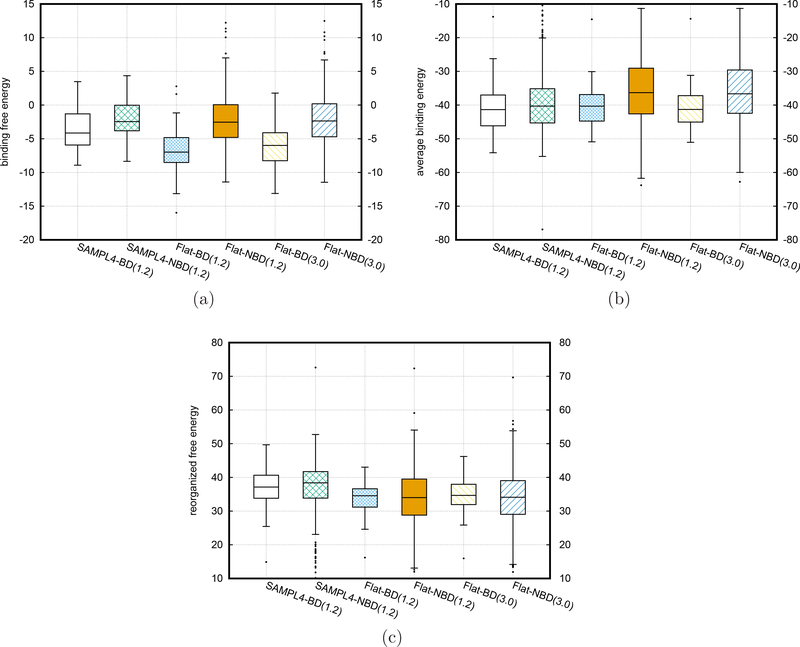
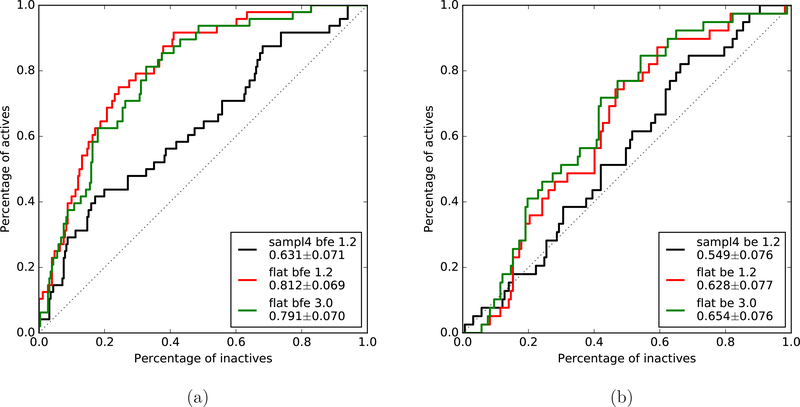
To accelerate conformation sampling of slow dynamics from receptor or ligand, we introduced flattening potentials on selected bonded and nonbonded intramolecular interactions to the binding energy distribution analysis method (BEDAM) for calculating absolute binding free energies of protein-ligand complexes using an implicit solvent model and implemented flattening BEDAM using the asynchronous replica exchange (AsyncRE) framework for performing large scale replica exchange molecular dynamics (REMD) simulations. The advantage of using the flattening feature to reduce high energy barriers was exhibited first by the p-xylene-T4 lysozyme complex, where the intramolecular interactions of a protein side chain on the binding site were flattened to accelerate the conformational transition of the side chain from the trans to the gauche state when the p-xylene ligand is present in the binding site. Much more extensive flattening BEDAM simulations were performed for 53 experimental binders and 248 nonbinders of HIV-1 integrase which formed the SAMPL4 challenge, with the total simulation time of 24.3 μs. We demonstrated that the flattening BEDAM simulations not only substantially increase the number of true positives (and reduce false negatives) but also improve the prediction accuracy of binding poses of experimental binders. Furthermore, the values of area under the curve (AUC) of receiver operating characteristic (ROC) and the enrichment factors at 20% cutoff calculated from the flattening BEDAM simulations were improved significantly in comparison with that of simulations without flattening as we previously reported for the whole SAMPL4 database. Detailed analysis found that the improved ability to discriminate the binding free energies between the binders and nonbinders is due to the fact that the flattening simulations reduce the reorganization free energy penalties of binders and decrease the overlap of binding free energy distributions of binders relative to that of nonbinders. This happens because the conformational ensemble distributions for both the ligand and protein in solution match those at the fully coupled (complex) state more closely when the systems are more fully sampled after the flattening potentials are applied to the intermediate states.
Figures




Similar articles
-
Massive-Scale Binding Free Energy Simulations of HIV Integrase Complexes Using Asynchronous Replica Exchange Framework Implemented on the IBM WCG Distributed Network.J Chem Inf Model. 2019 Apr 22;59(4):1382-1397. doi: 10.1021/acs.jcim.8b00817. Epub 2019 Feb 22. J Chem Inf Model. 2019. PMID: 30758197 Free PMC article.
-
Binding Energy Distribution Analysis Method: Hamiltonian Replica Exchange with Torsional Flattening for Binding Mode Prediction and Binding Free Energy Estimation.J Chem Theory Comput. 2016 May 10;12(5):2459-70. doi: 10.1021/acs.jctc.6b00134. Epub 2016 Apr 26. J Chem Theory Comput. 2016. PMID: 27070865 Free PMC article.
-
Large scale free energy calculations for blind predictions of protein-ligand binding: the D3R Grand Challenge 2015.J Comput Aided Mol Des. 2016 Sep;30(9):743-751. doi: 10.1007/s10822-016-9952-x. Epub 2016 Aug 25. J Comput Aided Mol Des. 2016. PMID: 27562018 Free PMC article.
-
Virtual screening of integrase inhibitors by large scale binding free energy calculations: the SAMPL4 challenge.J Comput Aided Mol Des. 2014 Apr;28(4):475-90. doi: 10.1007/s10822-014-9711-9. Epub 2014 Feb 7. J Comput Aided Mol Des. 2014. PMID: 24504704 Free PMC article.
-
Blind prediction of HIV integrase binding from the SAMPL4 challenge.J Comput Aided Mol Des. 2014 Apr;28(4):327-45. doi: 10.1007/s10822-014-9723-5. Epub 2014 Mar 5. J Comput Aided Mol Des. 2014. PMID: 24595873 Free PMC article. Review.
Cited by
-
Massive-Scale Binding Free Energy Simulations of HIV Integrase Complexes Using Asynchronous Replica Exchange Framework Implemented on the IBM WCG Distributed Network.J Chem Inf Model. 2019 Apr 22;59(4):1382-1397. doi: 10.1021/acs.jcim.8b00817. Epub 2019 Feb 22. J Chem Inf Model. 2019. PMID: 30758197 Free PMC article.
-
Structure-based virtual screening workflow to identify antivirals targeting HIV-1 capsid.J Comput Aided Mol Des. 2022 Mar;36(3):193-203. doi: 10.1007/s10822-022-00446-5. Epub 2022 Mar 9. J Comput Aided Mol Des. 2022. PMID: 35262811 Free PMC article.
-
Sampling Conformational Changes of Bound Ligands Using Nonequilibrium Candidate Monte Carlo and Molecular Dynamics.J Chem Theory Comput. 2020 Mar 10;16(3):1854-1865. doi: 10.1021/acs.jctc.9b01066. Epub 2020 Feb 24. J Chem Theory Comput. 2020. PMID: 32058713 Free PMC article.
References
-
- Gilson MK; Zhou H-X Calculation of Protein-Ligand Binding Affinities. Annu. Rev. Biophys. Biomol. Struct 2007, 36, 21–42. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
