

Two novel PRNP truncating mutations broaden the spectrum of prion amyloidosis
- PMID: 29928661
- PMCID: PMC5989776
- DOI: 10.1002/acn3.568
Two novel PRNP truncating mutations broaden the spectrum of prion amyloidosis
Abstract
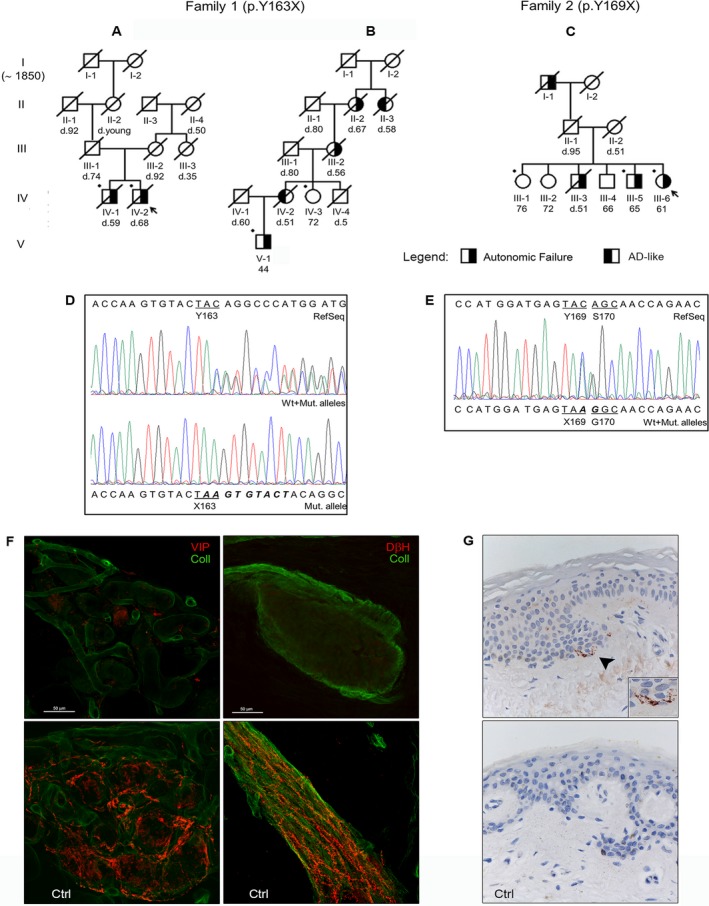
Truncating mutations in PRNP have been associated with heterogeneous phenotypes ranging from chronic diarrhea and neuropathy to dementia, either rapidly or slowly progressive. We identified novel PRNP stop-codon mutations (p.Y163X, p.Y169X) in two Italian kindreds. Disease typically presented in the third or fourth decade with progressive autonomic failure and diarrhea. Moreover, one proband (p.Y163X) developed late cognitive decline, whereas some of his relatives presented with isolated cognitive and psychiatric symptoms. Our results strengthen the link between PRNP truncating mutations and systemic abnormal PrP deposition and support a wider application of PRNP screening to include unsolved cases of familial autonomic neuropathy.
Figures
References
-
- Parchi P, Saverioni D. Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol 2012;50:20–45. - PubMed
-
- Ghetti B, Tagliavini F, Takao M, et al. Hereditary prion protein amyloidoses. Clin Lab Med 2003;23:65–85. - PubMed
-
- Stahl N, Baldwin MA, Burlingame AL, et al. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 1990;29:8879–8884. - PubMed
-
- Kitamoto T, Iizuka R, Tateishi J. An amber mutation of prion protein in gerstmann‐straussler syndrome with mutant PrP plaques. Biochem Biophys Res Commun 1993;192:525–531. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
