

Gene length corrected trimmed mean of M-values (GeTMM) processing of RNA-seq data performs similarly in intersample analyses while improving intrasample comparisons
- PMID: 29929481
- PMCID: PMC6013957
- DOI: 10.1186/s12859-018-2246-7
Gene length corrected trimmed mean of M-values (GeTMM) processing of RNA-seq data performs similarly in intersample analyses while improving intrasample comparisons
Abstract
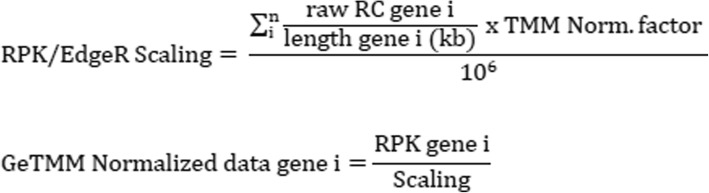
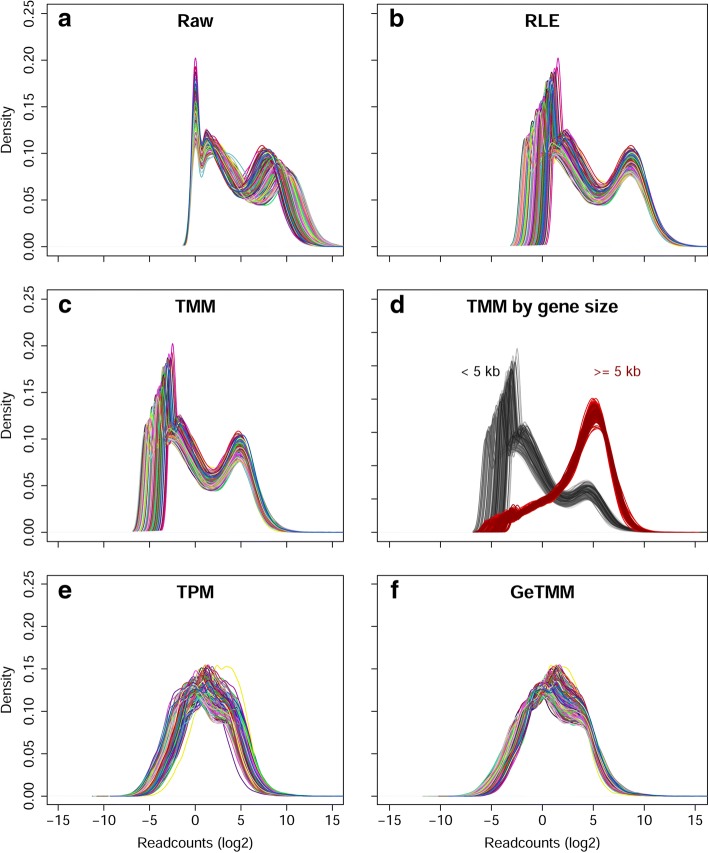
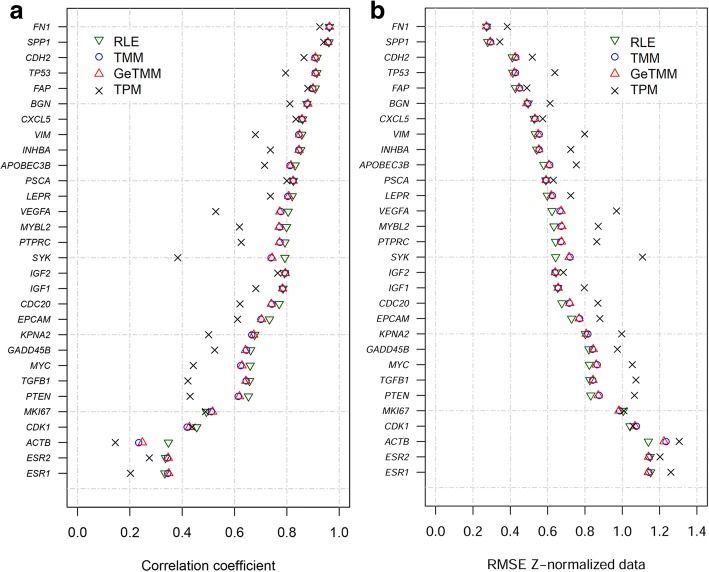
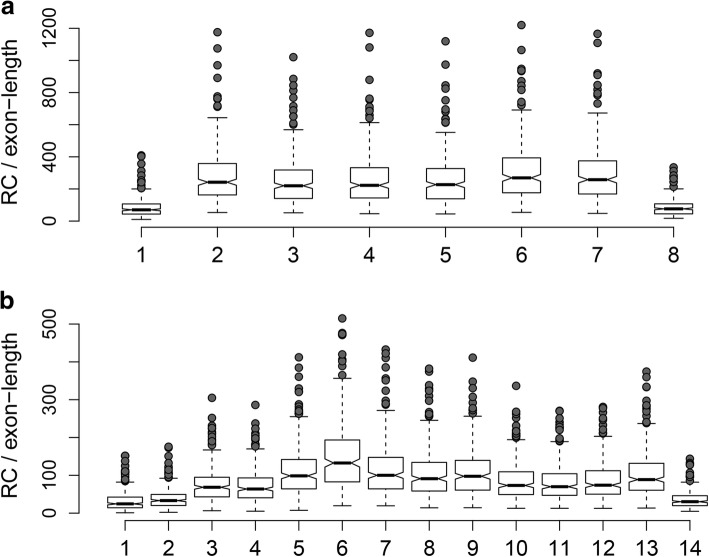
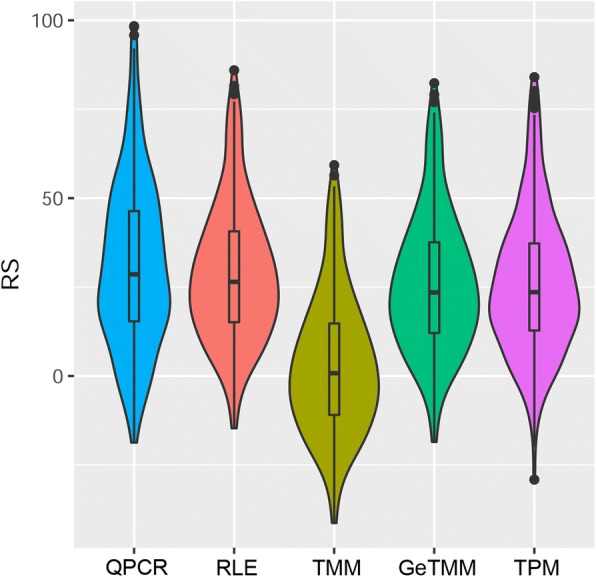
Background: Current normalization methods for RNA-sequencing data allow either for intersample comparison to identify differentially expressed (DE) genes or for intrasample comparison for the discovery and validation of gene signatures. Most studies on optimization of normalization methods typically use simulated data to validate methodologies. We describe a new method, GeTMM, which allows for both inter- and intrasample analyses with the same normalized data set. We used actual (i.e. not simulated) RNA-seq data from 263 colon cancers (no biological replicates) and used the same read count data to compare GeTMM with the most commonly used normalization methods (i.e. TMM (used by edgeR), RLE (used by DESeq2) and TPM) with respect to distributions, effect of RNA quality, subtype-classification, recurrence score, recall of DE genes and correlation to RT-qPCR data.
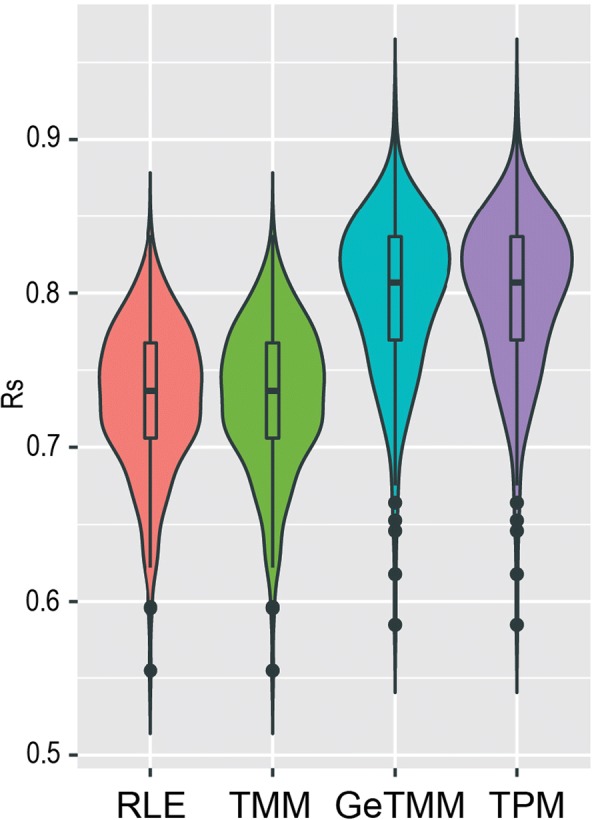
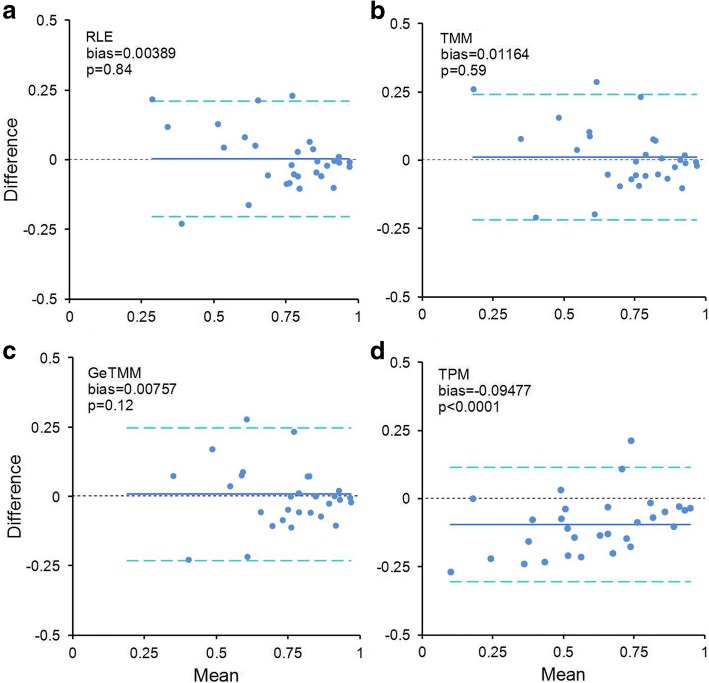
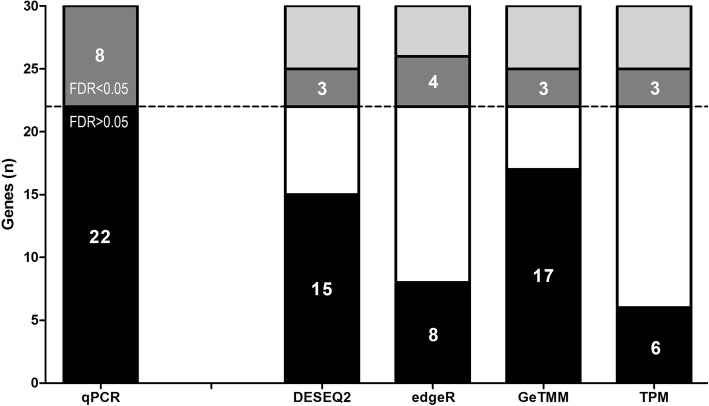
Results: We observed a clear benefit for GeTMM and TPM with regard to intrasample comparison while GeTMM performed similar to TMM and RLE normalized data in intersample comparisons. Regarding DE genes, recall was found comparable among the normalization methods, while GeTMM showed the lowest number of false-positive DE genes. Remarkably, we observed limited detrimental effects in samples with low RNA quality.
Conclusions: We show that GeTMM outperforms established methods with regard to intrasample comparison while performing equivalent with regard to intersample normalization using the same normalized data. These combined properties enhance the general usefulness of RNA-seq but also the comparability to the many array-based gene expression data in the public domain.
Keywords: Colorectal Cancer; DESeq2; GeTMM; Normalization methods; RNA sequencing; TPM; edgeR.
Conflict of interest statement
Ethics approval and consent to participate
All patients gave written informed consent for the collection and use of both clinical data and tumor tissue (Institutional Review Board Erasmus MC University Medical Center; MEC-2007-088).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
