

Extending the ophthalmological phenotype of Galloway-Mowat syndrome with distinct retinal dysfunction: a report and review of ocular findings
- PMID: 29929488
- PMCID: PMC6013877
- DOI: 10.1186/s12886-018-0820-4
Extending the ophthalmological phenotype of Galloway-Mowat syndrome with distinct retinal dysfunction: a report and review of ocular findings
Abstract
Background: Galloway-Mowat syndrome (GMS) is a rare autosomal recessive condition first described in 1968 and characterized by microcephaly and infantile onset of central nervous system (CNS) abnormalities resulting in severely delayed psychomotor development, cerebellar atrophy, epilepsy, and ataxia, as well as renal abnormalities such as nephrotic syndrome, proteinuria, end-stage renal disease (ESRD), and hiatal hernia.
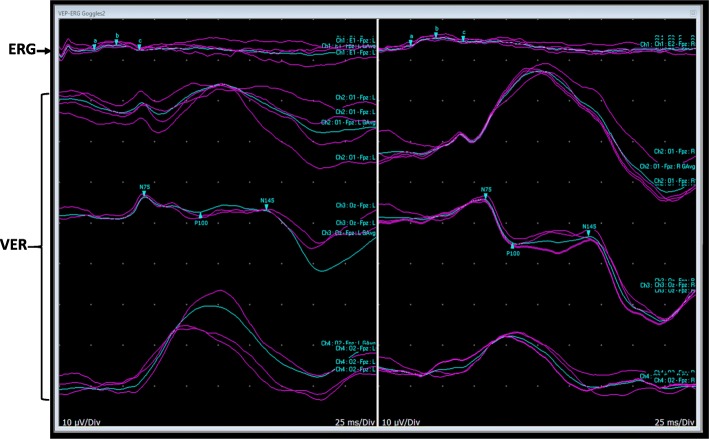
Case presentation: We describe a GMS case diagnosed with homozygous missense mutation in the WDR73 gene, with absence of renal abnormalities. We expanded the clinical phenotype of GMS with WDR73 gene defect to include retinal dysfunction with missense mutation and developmental dysplasia of the hip. We compared eye findings of our case to previously reported cases, and we present an electroretinogram (ERG) picture for the first time in the literature.
Conclusion: We recommend that clinicians screen patients with GM syndrome for retinal dysfunction and that a skeletal survey should be done to detect developmental dysplasia of the hip (DDH) so as to provide for early intervention.
Keywords: Absence of perinatal and neonatal renal dysfunction; Galloway-Mowat syndrome; Retinal dysfunction; WDR37.
Conflict of interest statement
Ethics approval and consent to participate
This study was approved by the Institutional Review Board at King Abdullah International Medical Research Centre (KIMARC) (Study number: RC16/113/R).
Consent for publication
Written informed consent was obtained from the parents.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Delague V, Bareil C, Bouvagnet P, et al. A new autosomal recessive non-progressive congenital cerebellar ataxia associated with mental retardation, optic atrophy, and skin abnormalities (CAMOS) maps to chromosome 15q24-q26 in a large consanguineous Lebanese Druze family. Neurogenetics. 2002;4(1):23–27. doi: 10.1007/s10048-001-0127-z. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
