IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes
- PMID: 29930244
- PMCID: PMC6013502
- DOI: 10.1038/s41467-018-04365-8
IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes
Abstract
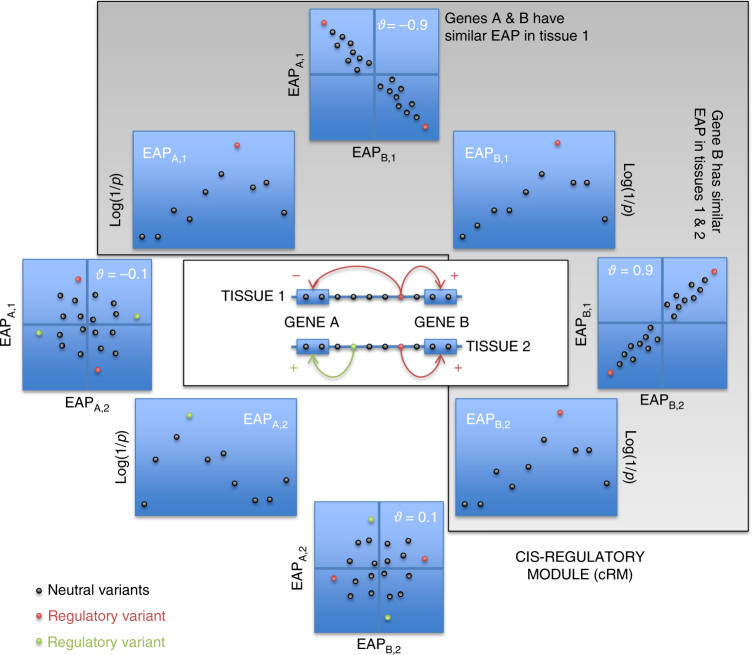
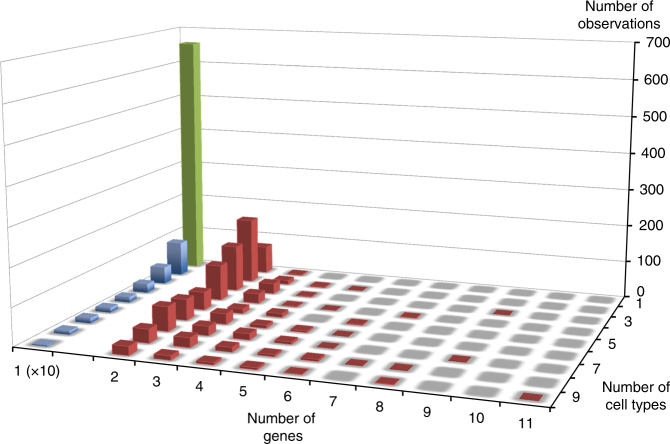
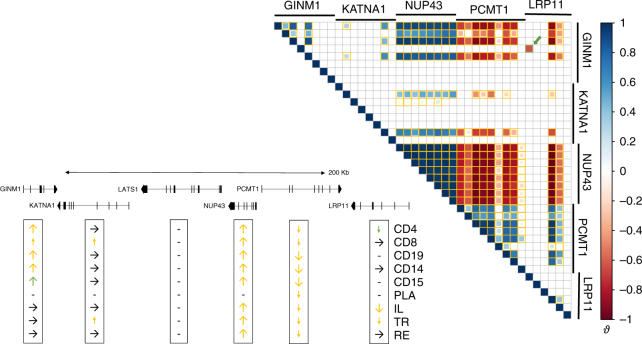
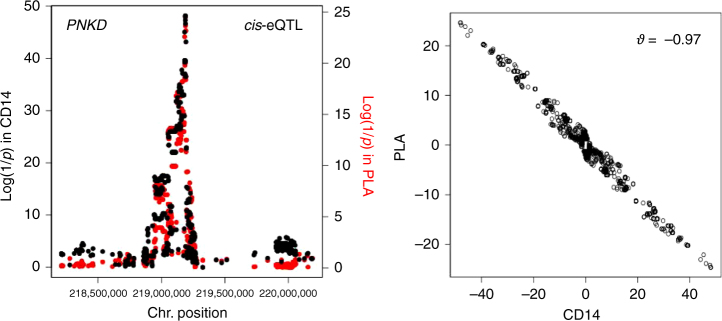
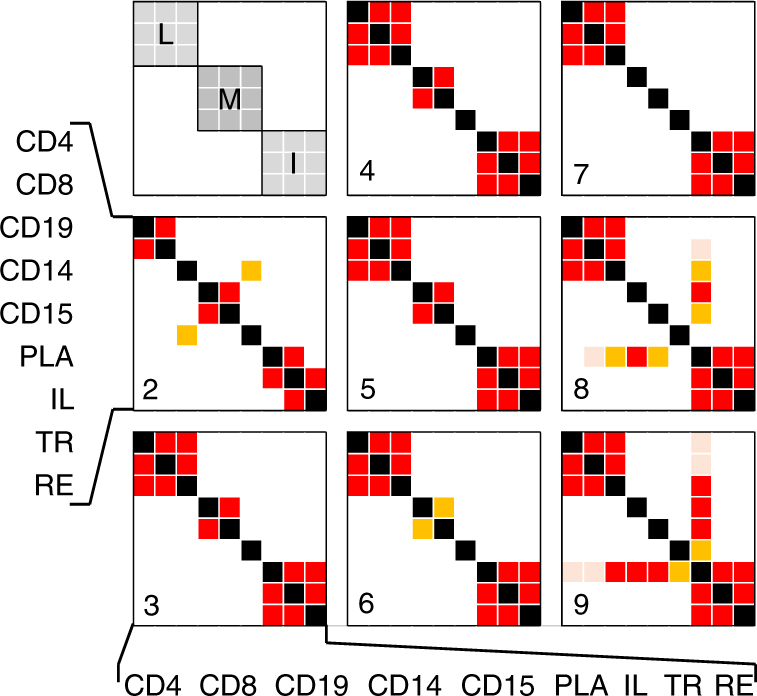
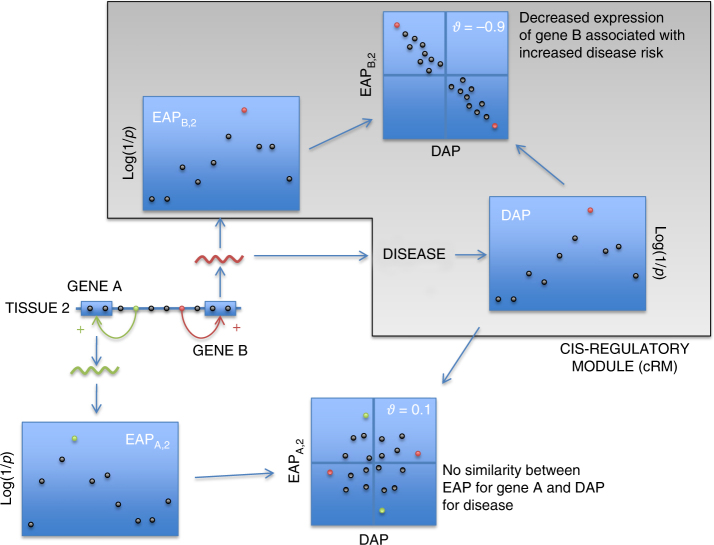
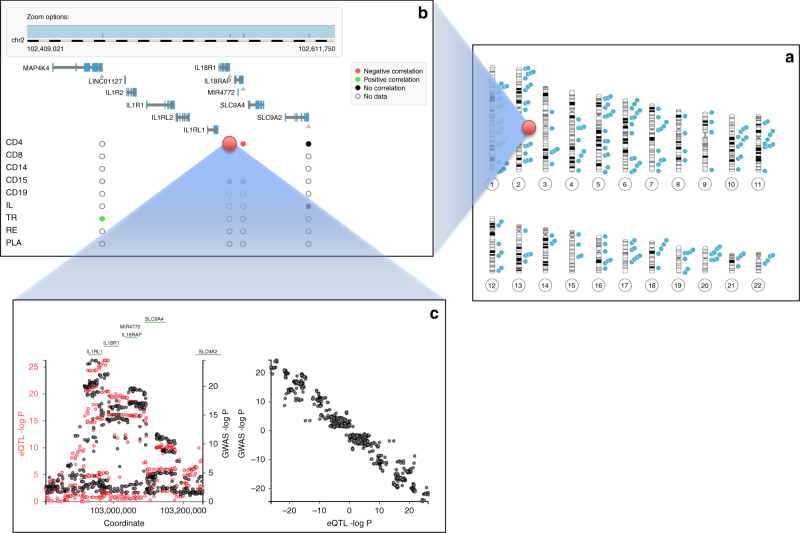
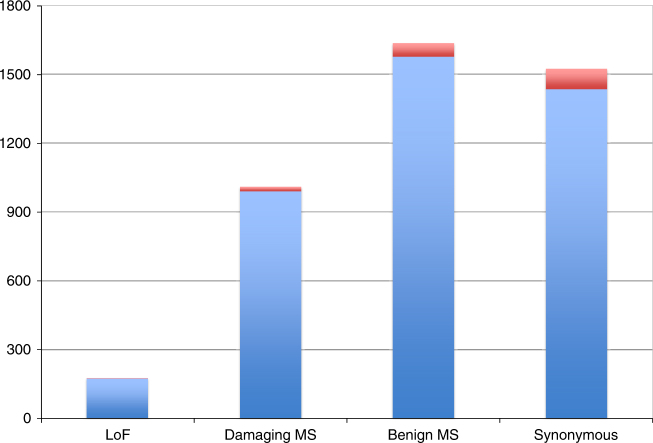
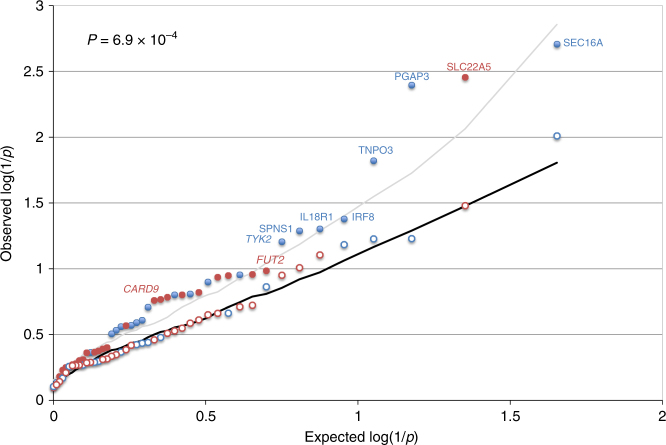
GWAS have identified >200 risk loci for Inflammatory Bowel Disease (IBD). The majority of disease associations are known to be driven by regulatory variants. To identify the putative causative genes that are perturbed by these variants, we generate a large transcriptome data set (nine disease-relevant cell types) and identify 23,650 cis-eQTL. We show that these are determined by ∼9720 regulatory modules, of which ∼3000 operate in multiple tissues and ∼970 on multiple genes. We identify regulatory modules that drive the disease association for 63 of the 200 risk loci, and show that these are enriched in multigenic modules. Based on these analyses, we resequence 45 of the corresponding 100 candidate genes in 6600 Crohn disease (CD) cases and 5500 controls, and show with burden tests that they include likely causative genes. Our analyses indicate that ≥10-fold larger sample sizes will be required to demonstrate the causality of individual genes using this approach.
Conflict of interest statement
The authors declare no competing interests.
Figures