

Recent examples of α-ketoglutarate-dependent mononuclear non-haem iron enzymes in natural product biosyntheses
- PMID: 29932179
- PMCID: PMC6093783
- DOI: 10.1039/c7np00067g
Recent examples of α-ketoglutarate-dependent mononuclear non-haem iron enzymes in natural product biosyntheses
Abstract
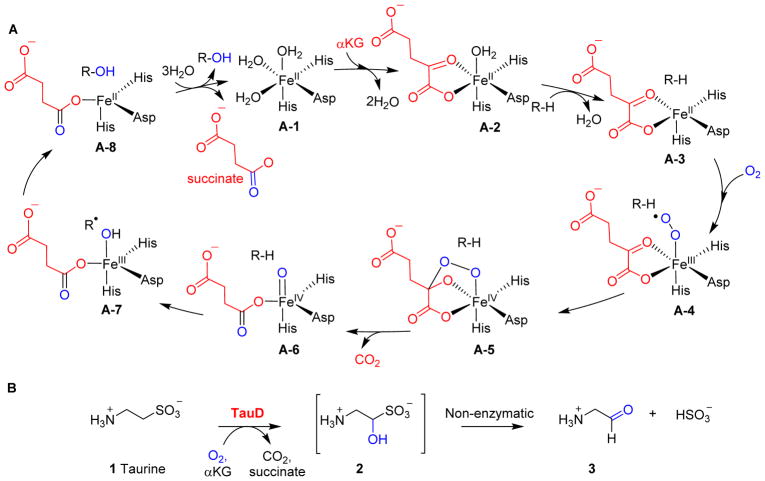
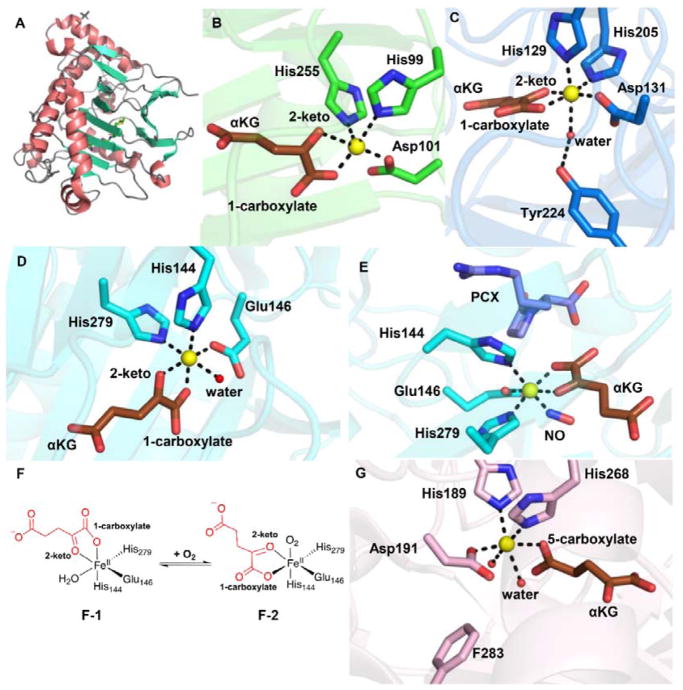
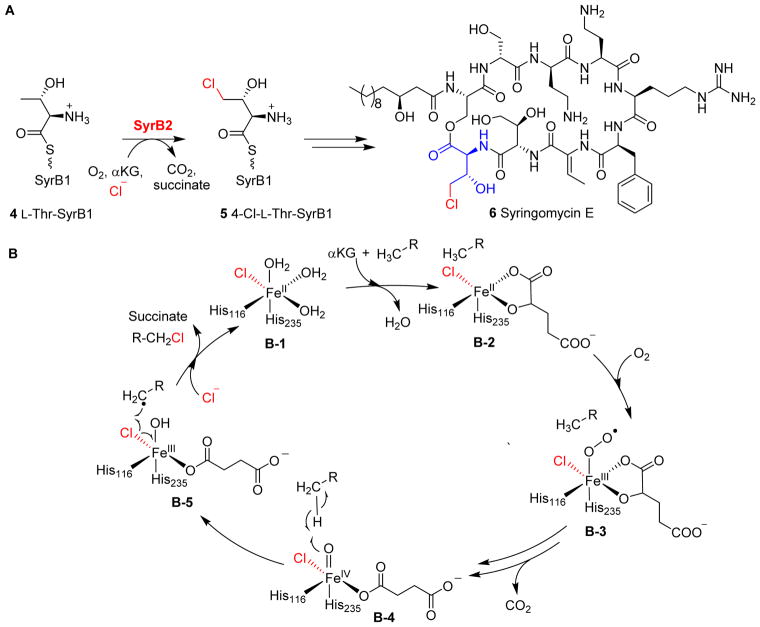
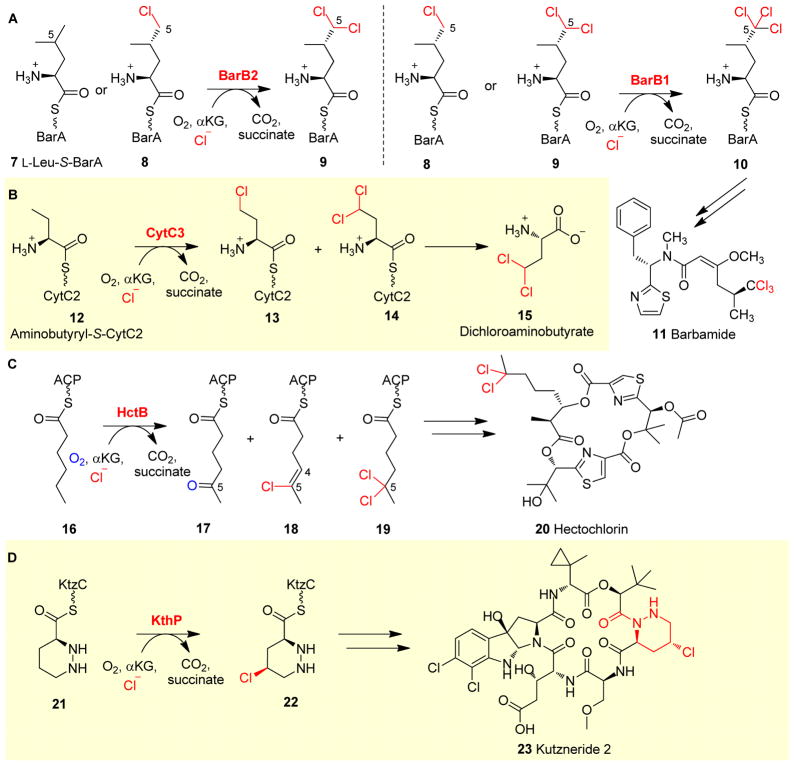
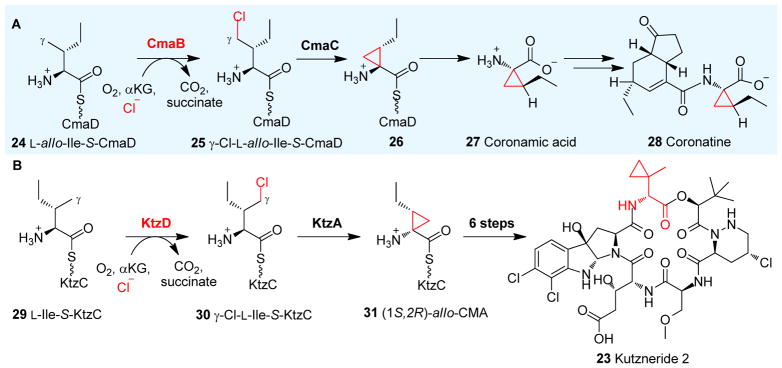
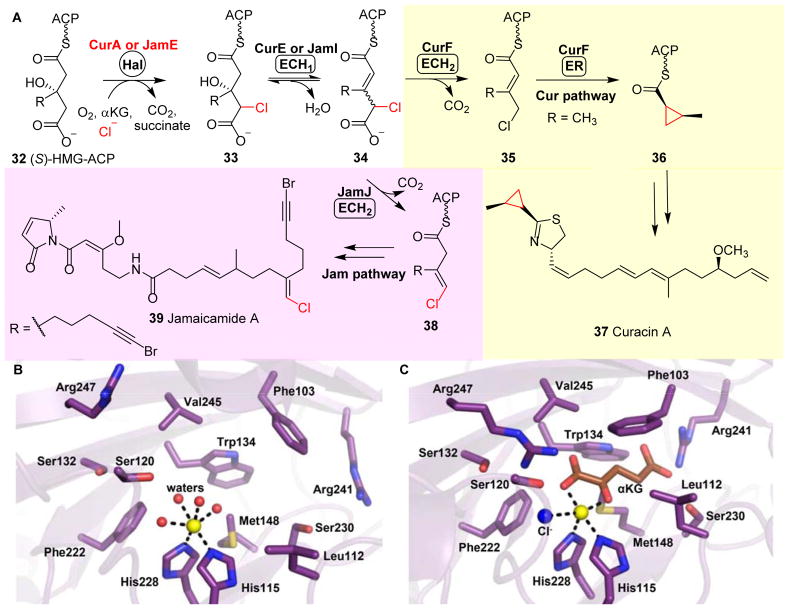
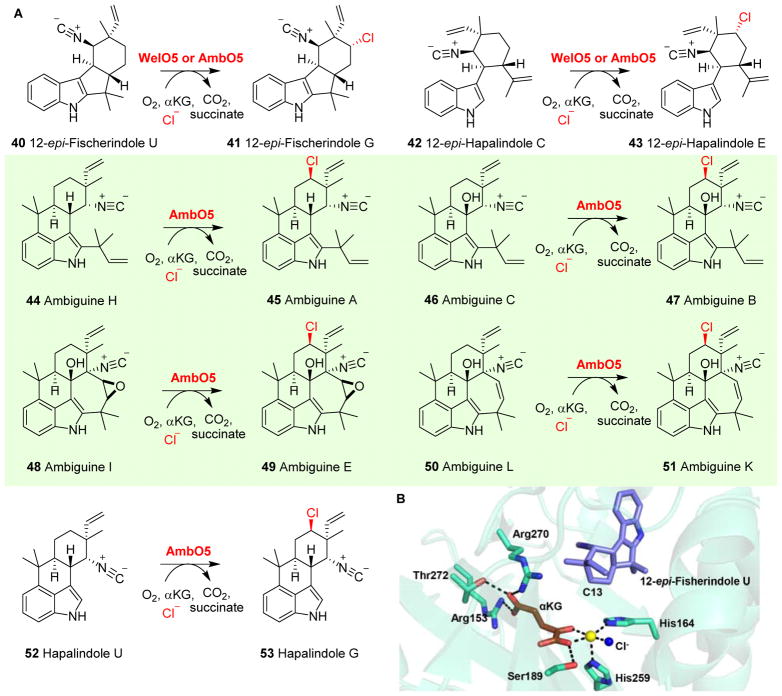
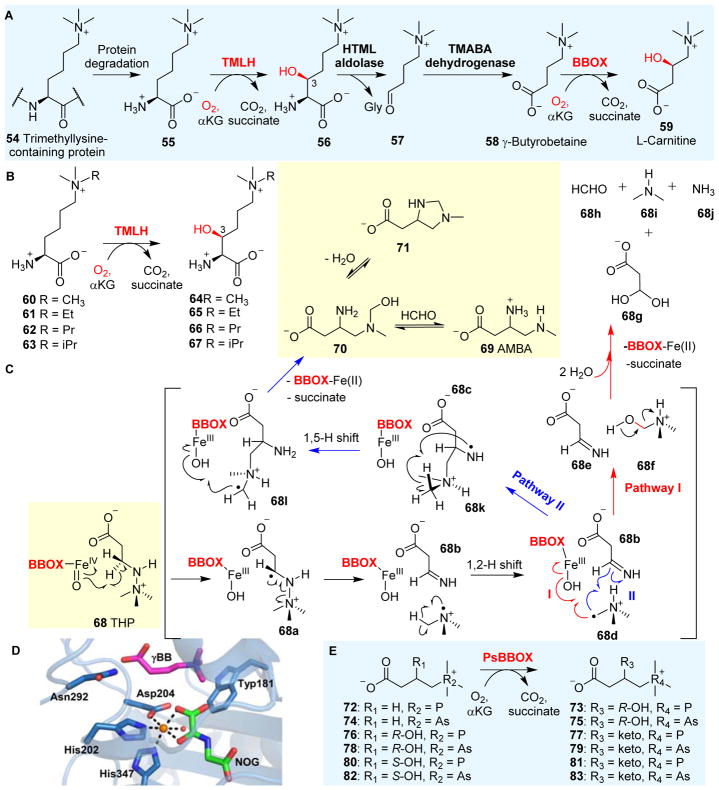
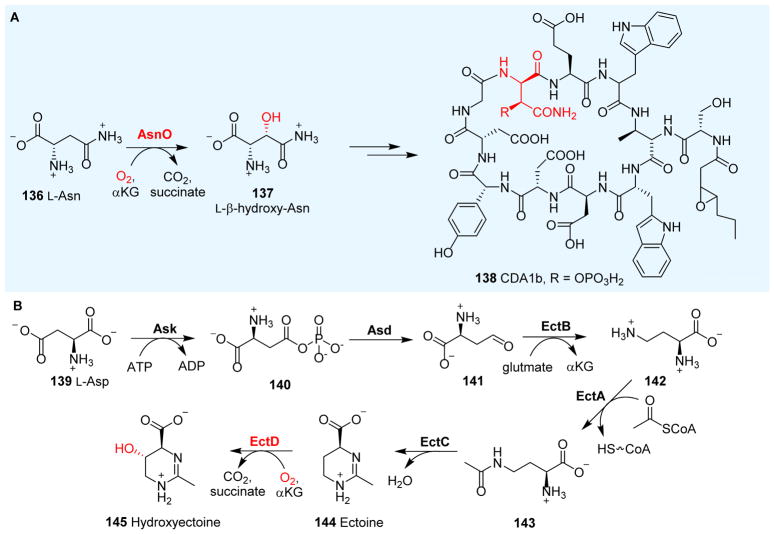
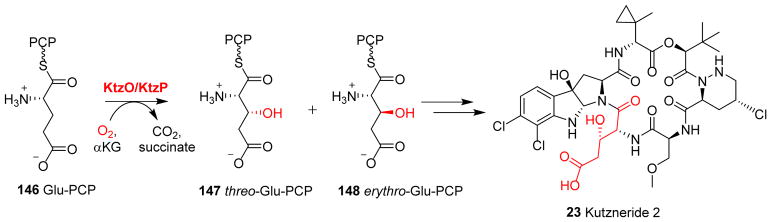
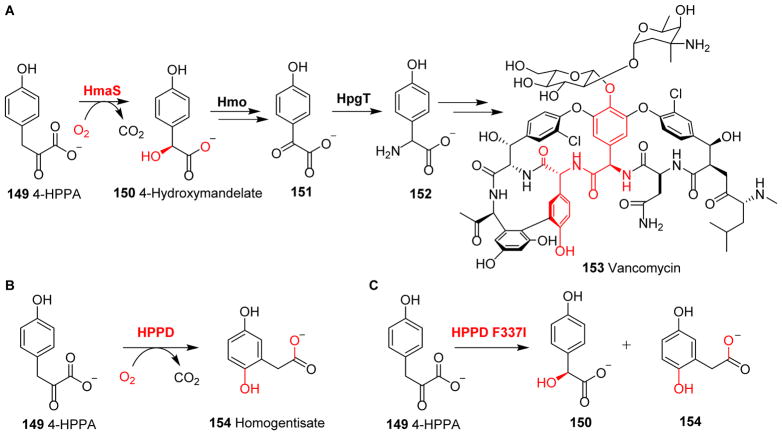
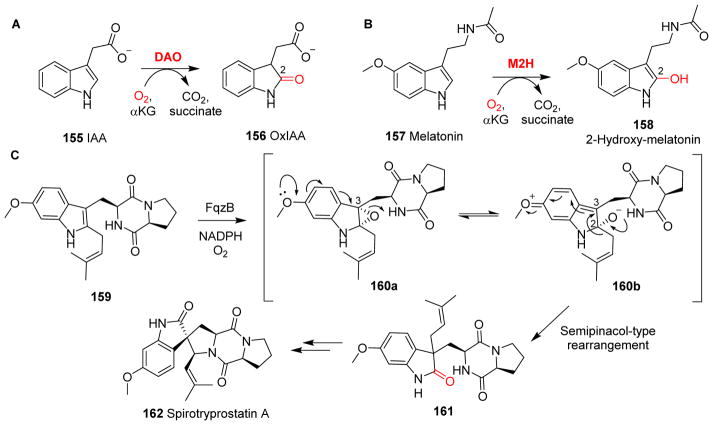
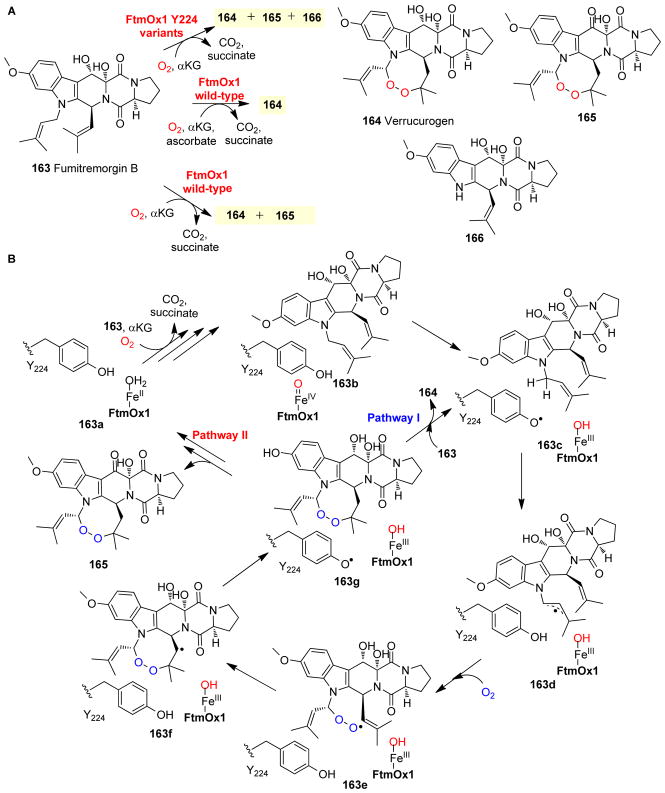
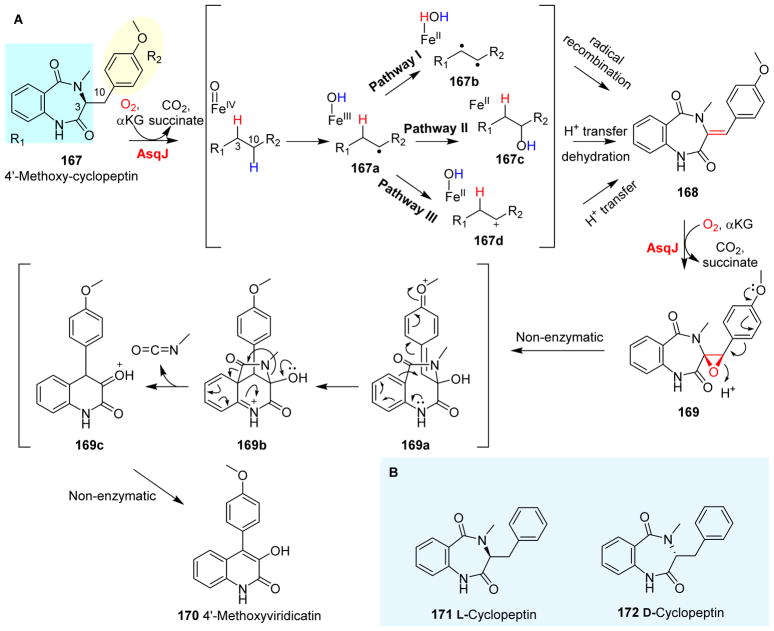
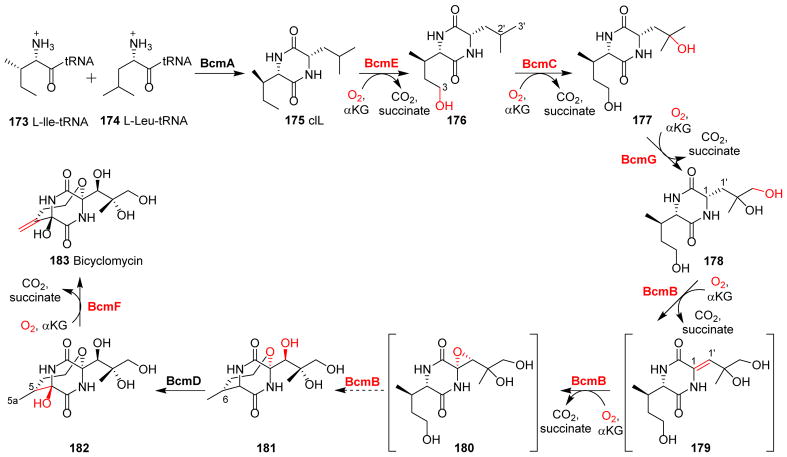
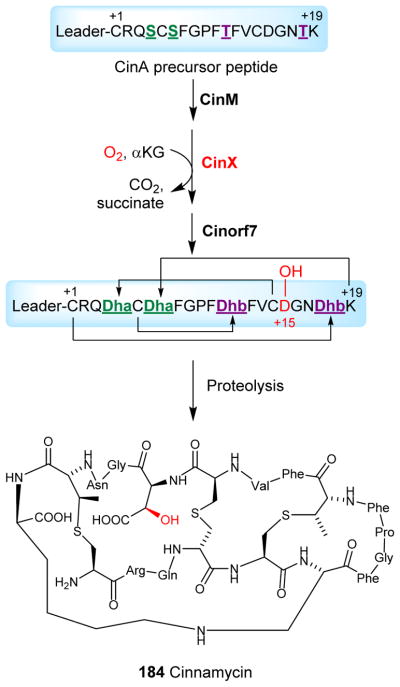
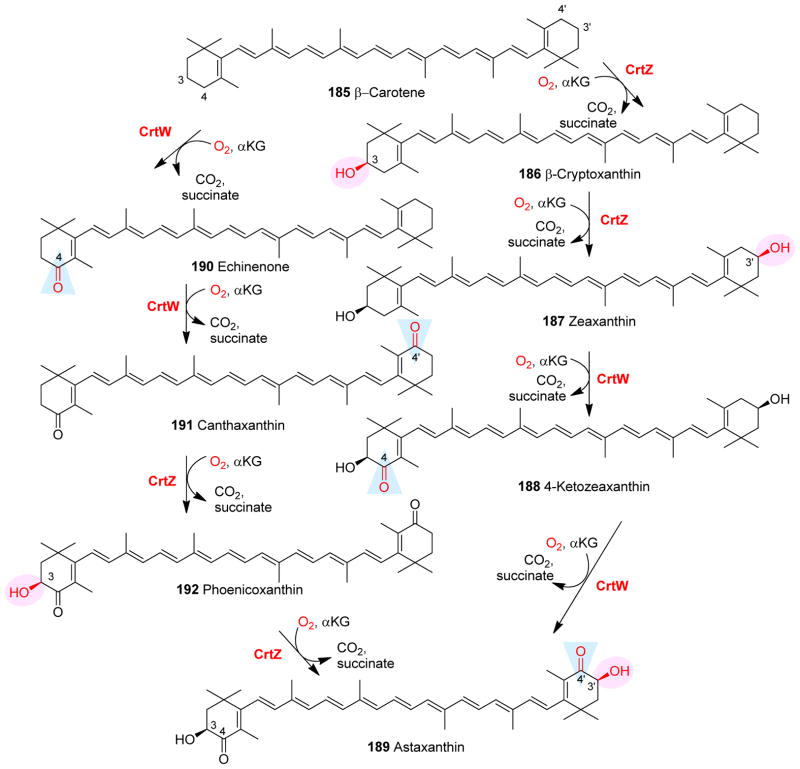
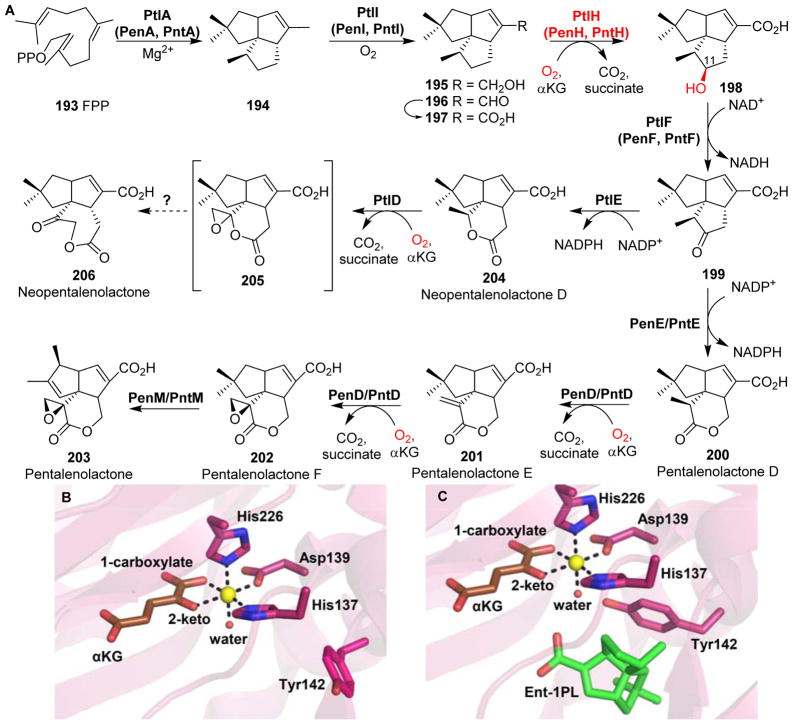
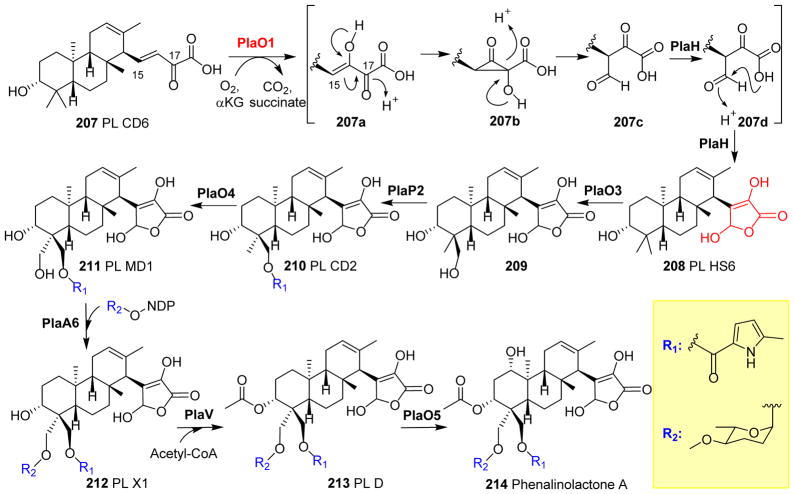
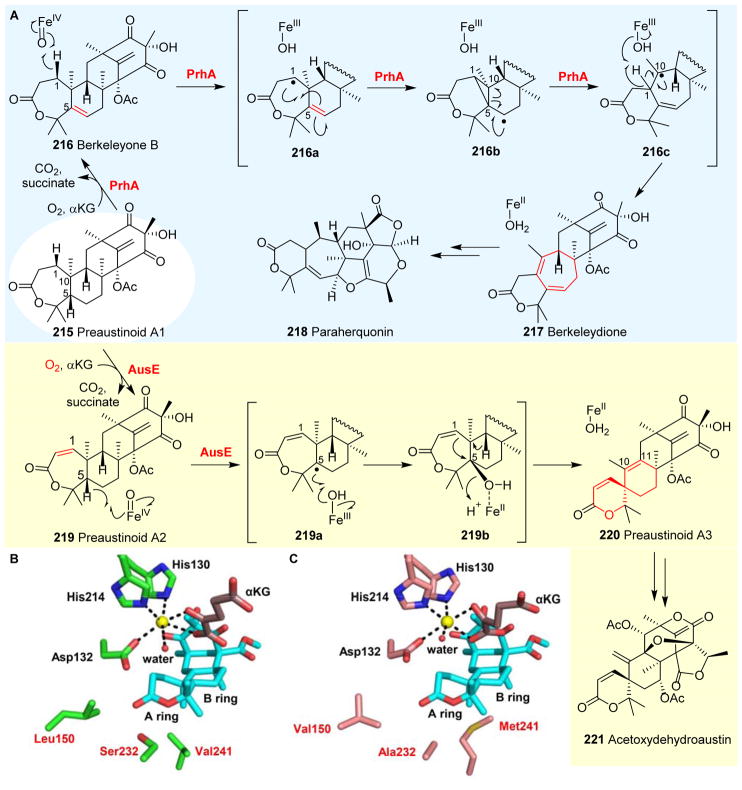
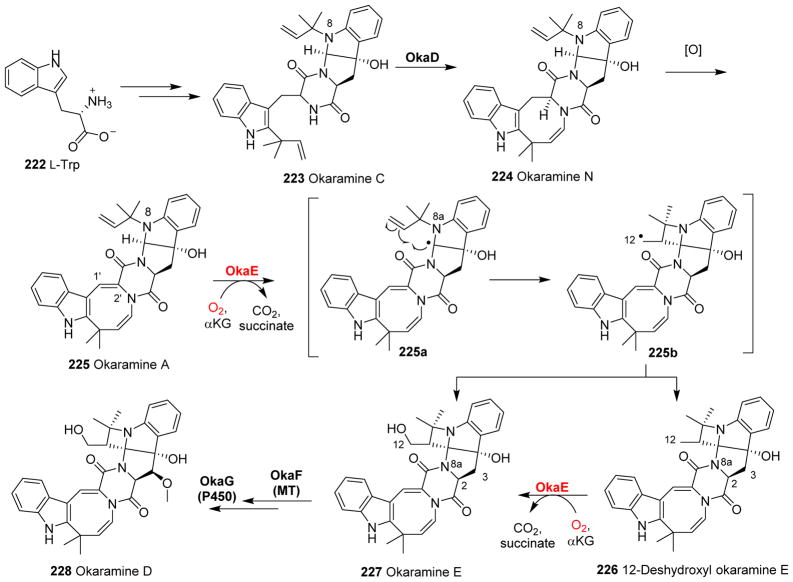
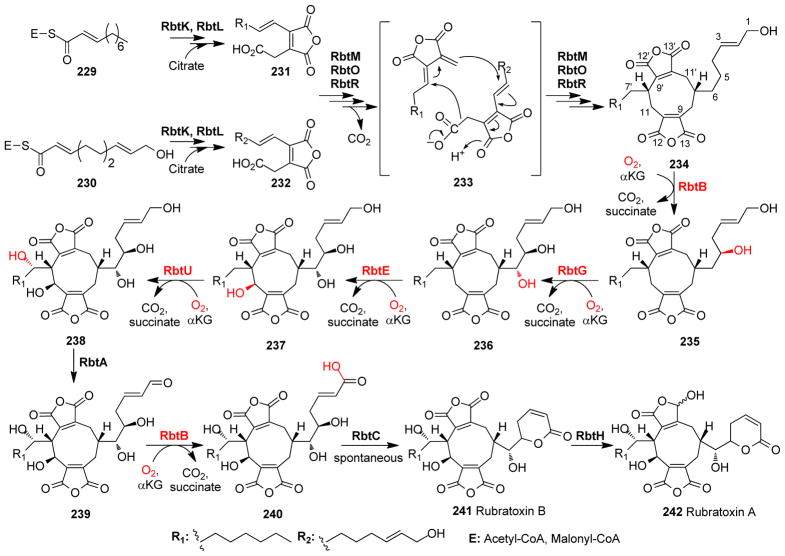
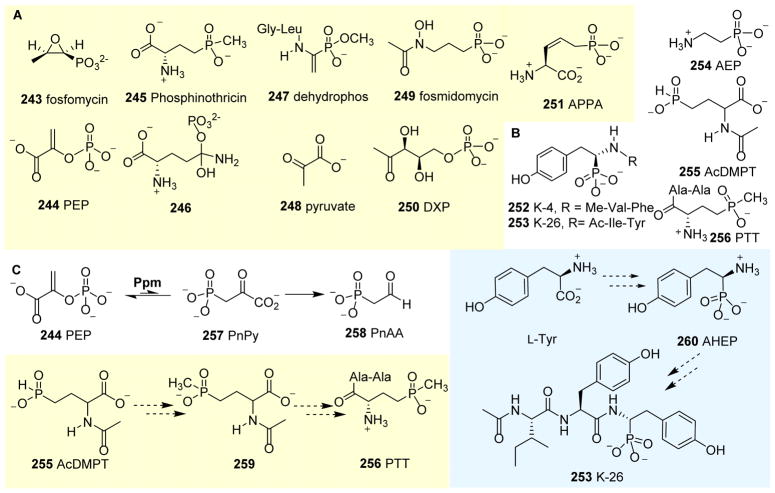
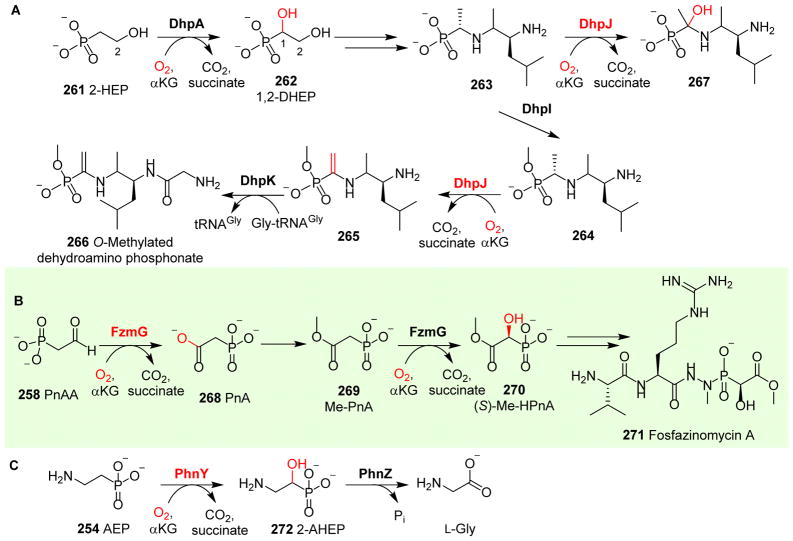
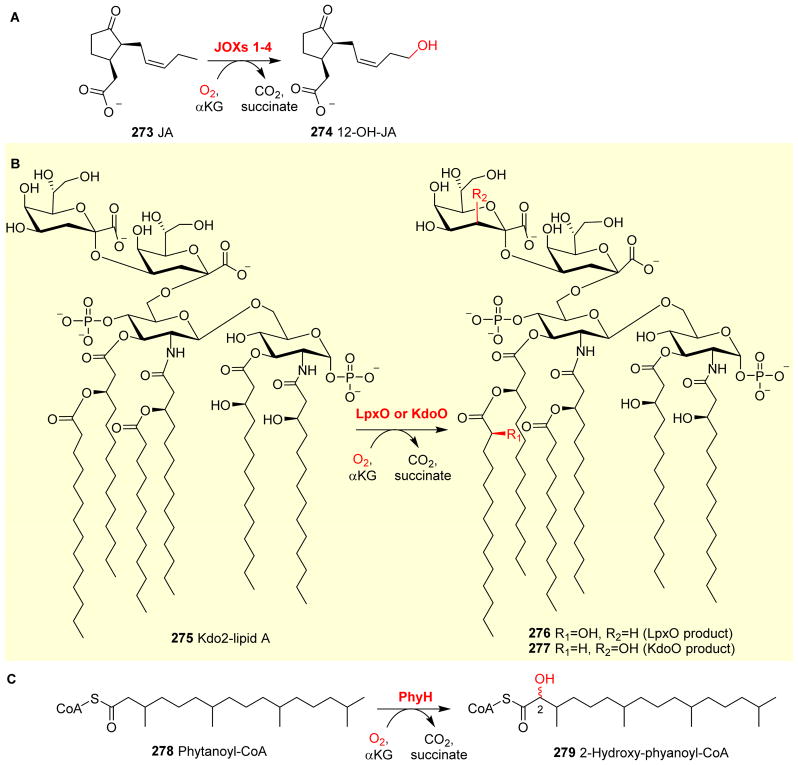
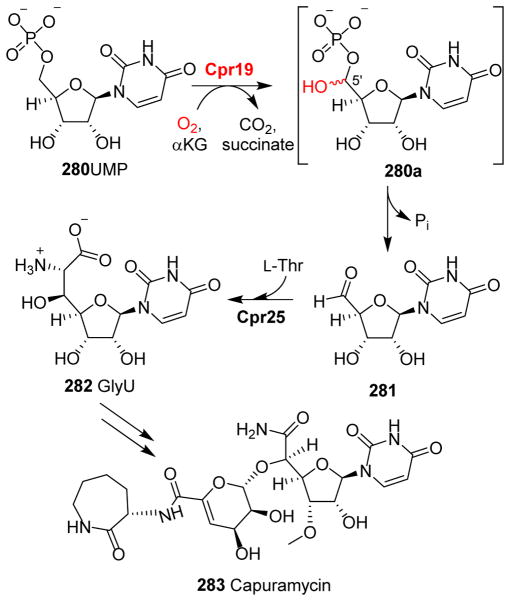
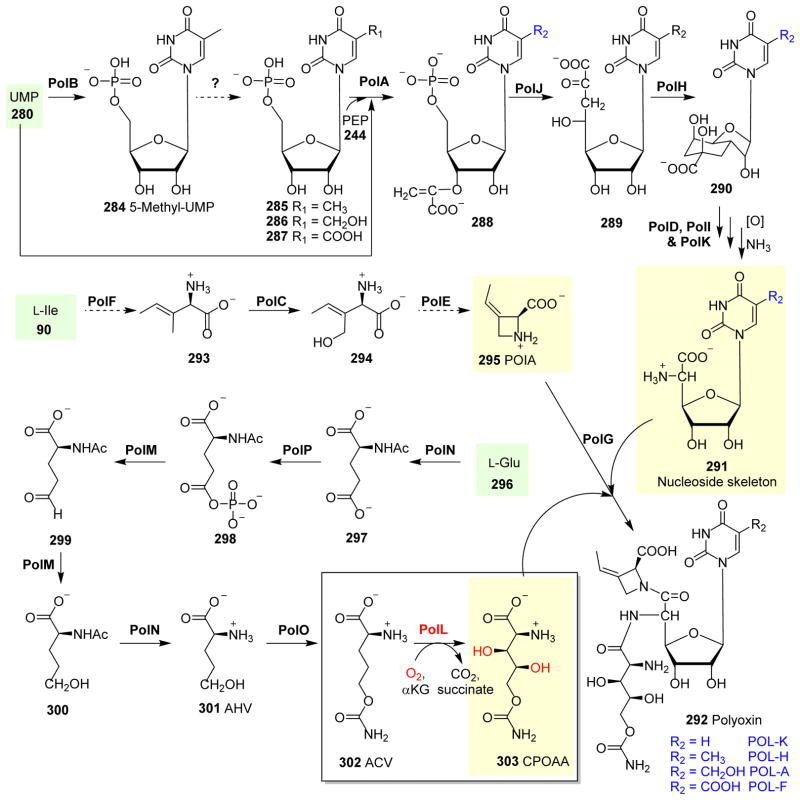
Covering: up to 2018 α-Ketoglutarate (αKG, also known as 2-oxoglutarate)-dependent mononuclear non-haem iron (αKG-NHFe) enzymes catalyze a wide range of biochemical reactions, including hydroxylation, ring fragmentation, C-C bond cleavage, epimerization, desaturation, endoperoxidation and heterocycle formation. These enzymes utilize iron(ii) as the metallo-cofactor and αKG as the co-substrate. Herein, we summarize several novel αKG-NHFe enzymes involved in natural product biosyntheses discovered in recent years, including halogenation reactions, amino acid modifications and tailoring reactions in the biosynthesis of terpenes, lipids, fatty acids and phosphonates. We also conducted a survey of the currently available structures of αKG-NHFe enzymes, in which αKG binds to the metallo-centre bidentately through either a proximal- or distal-type binding mode. Future structure-function and structure-reactivity relationship investigations will provide crucial information regarding how activities in this large class of enzymes have been fine-tuned in nature.
Conflict of interest statement
There are no conflicts to declare.
Figures
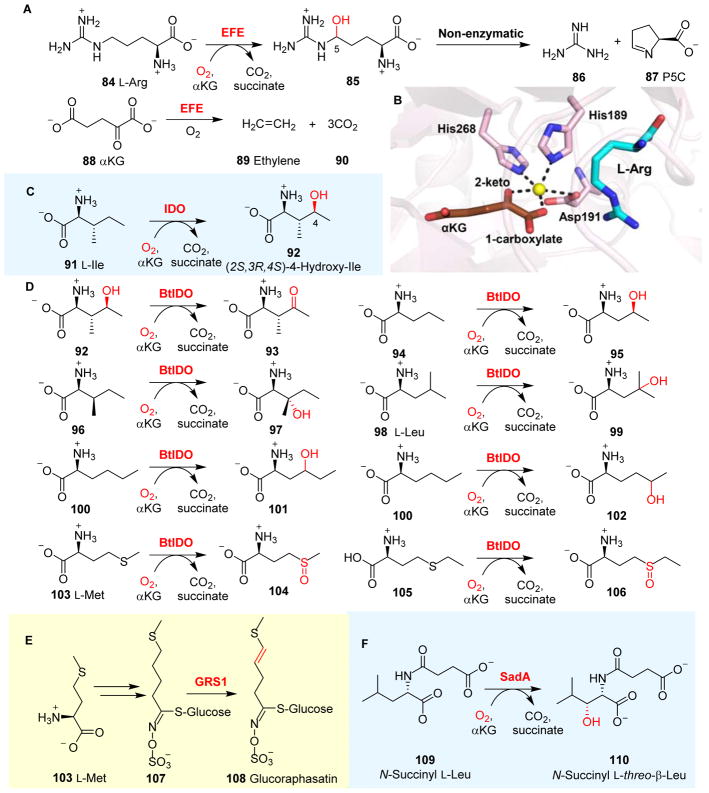
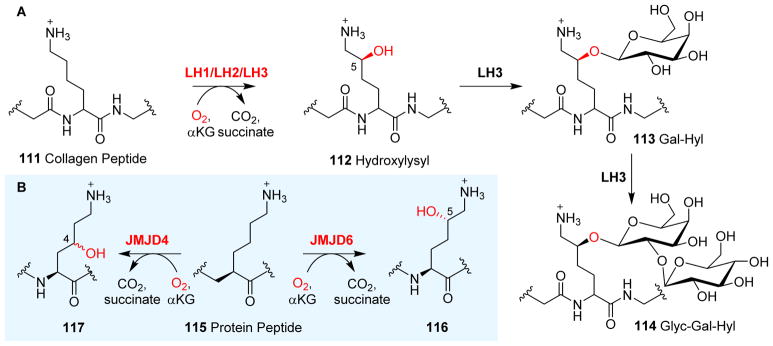
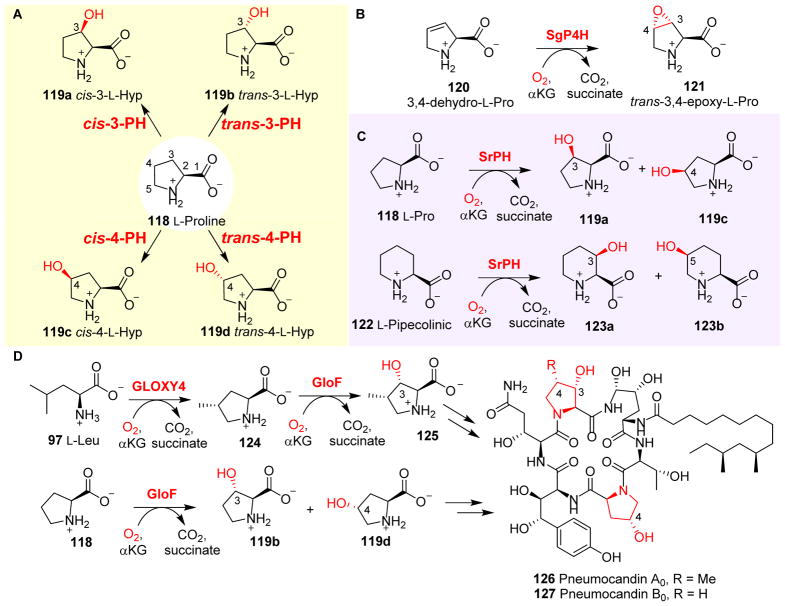
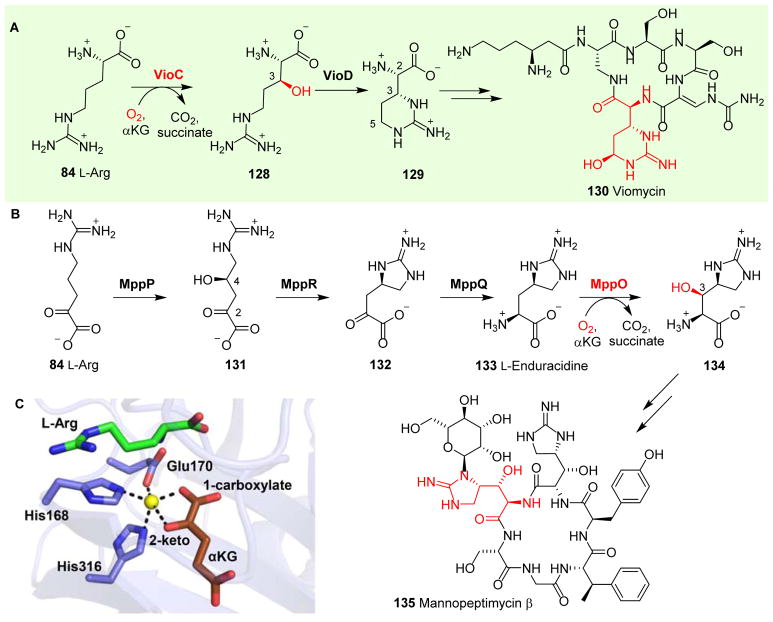
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical