

Novel therapies for combating chronic neuropathological sequelae of TBI
- PMID: 29933008
- PMCID: PMC6513317
- DOI: 10.1016/j.neuropharm.2018.06.021
Novel therapies for combating chronic neuropathological sequelae of TBI
Abstract
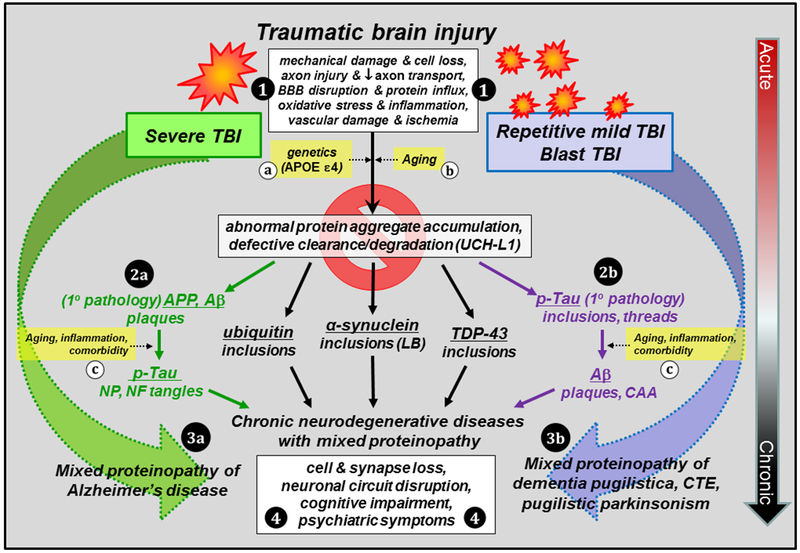
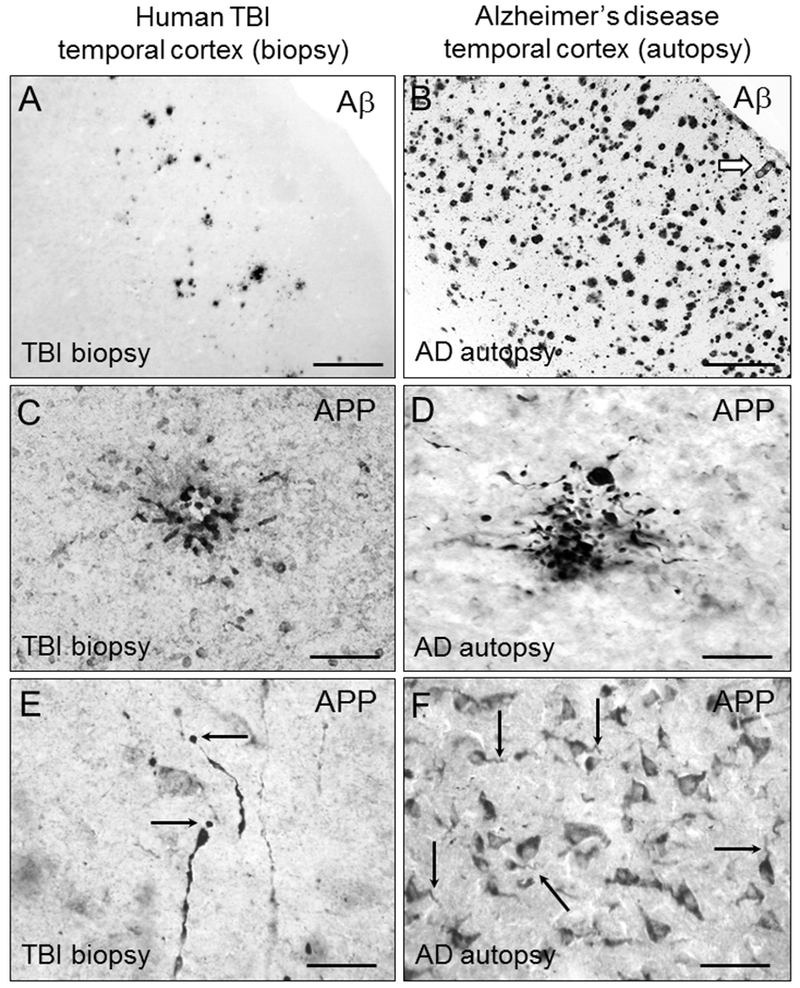
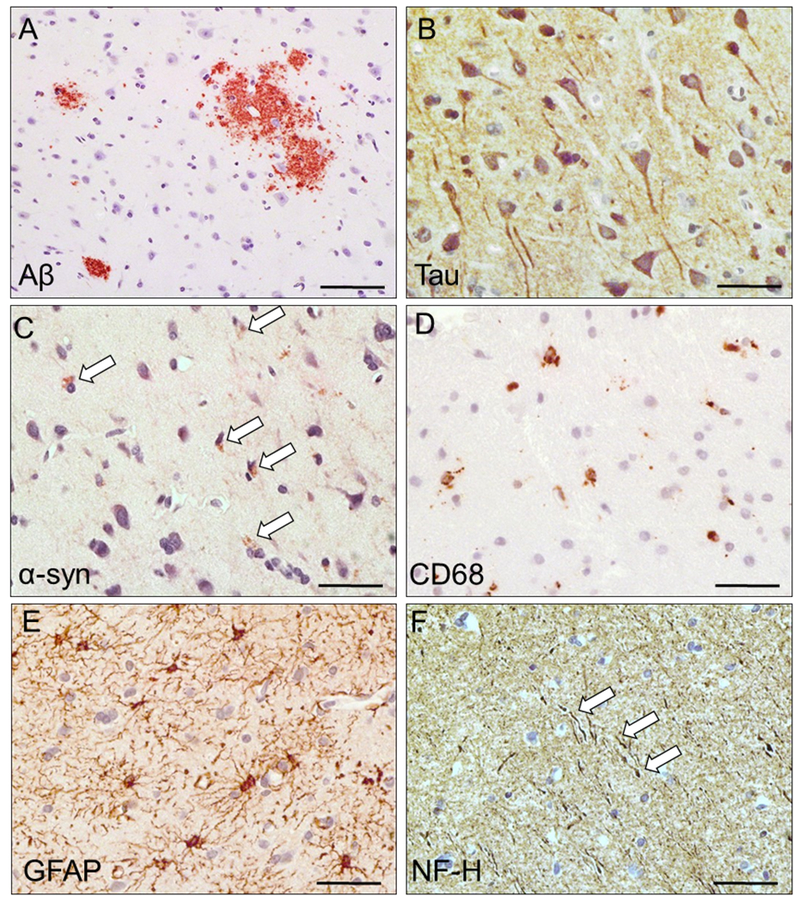
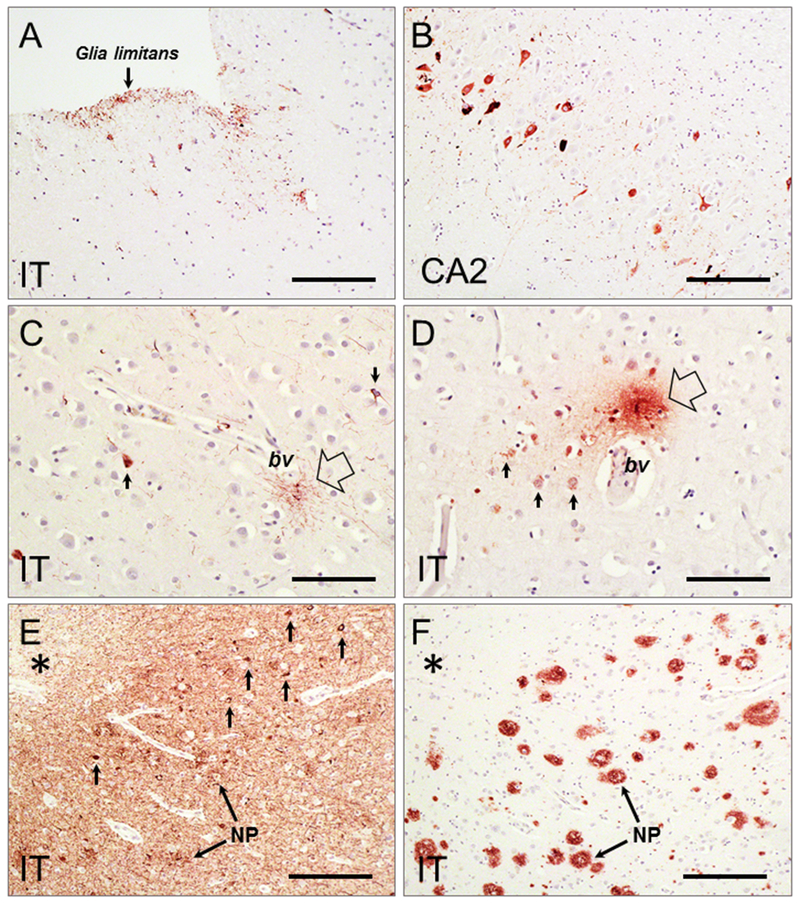
Traumatic brain injury (TBI) is a risk factor for development of chronic neurodegenerative disorders later in life. This review summarizes the current knowledge and concepts regarding the connection between long-term consequences of TBI and aging-associated neurodegenerative disorders including Alzheimer's disease (AD), chronic traumatic encephalopathy (CTE), and Parkinsonism, with implications for novel therapy targets. Several aggregation-prone proteins such as the amyloid-beta (Aβ) peptides, tau proteins, and α-synuclein protein are involved in secondary pathogenic cascades initiated by a TBI and are also major building blocks of the hallmark pathological lesions in chronic human neurodegenerative diseases with dementia. Impaired metabolism and degradation pathways of aggregation-prone proteins are discussed as potentially critical links between the long-term aftermath of TBI and chronic neurodegeneration. Utility and limitations of previous and current preclinical TBI models designed to study the link between TBI and chronic neurodegeneration, and promising intervention pharmacotherapies and non-pharmacologic strategies to break this link, are also summarized. Complexity of long-term neuropathological consequences of TBI is discussed, with a goal of guiding future preclinical studies and accelerating implementation of promising therapeutics into clinical trials. This article is part of the Special Issue entitled "Novel Treatments for Traumatic Brain Injury".
Keywords: Alpha synuclein; Alzheimer's disease; Amyloid; Brain trauma; Chronic traumatic encephalopathy; Tau.
Copyright © 2018. Published by Elsevier Ltd.
Figures
References
-
- Abisambra JF, Scheff S, 2014. Brain injury in the context of tauopathies. J Alzheimers Dis 40, 495–518. - PubMed
-
- Abrahamson EE, Ikonomovic MD, Ciallella JR, Hope CE, Paljug WR, Isanski BA, Flood DG, Clark RSB, DeKosky ST, 2006. Caspase inhibition therapy abolishes brain trauma-induced increases in Abeta peptide: Implications for clinical outcome. Exp Neurol 197, 437–450. - PubMed
-
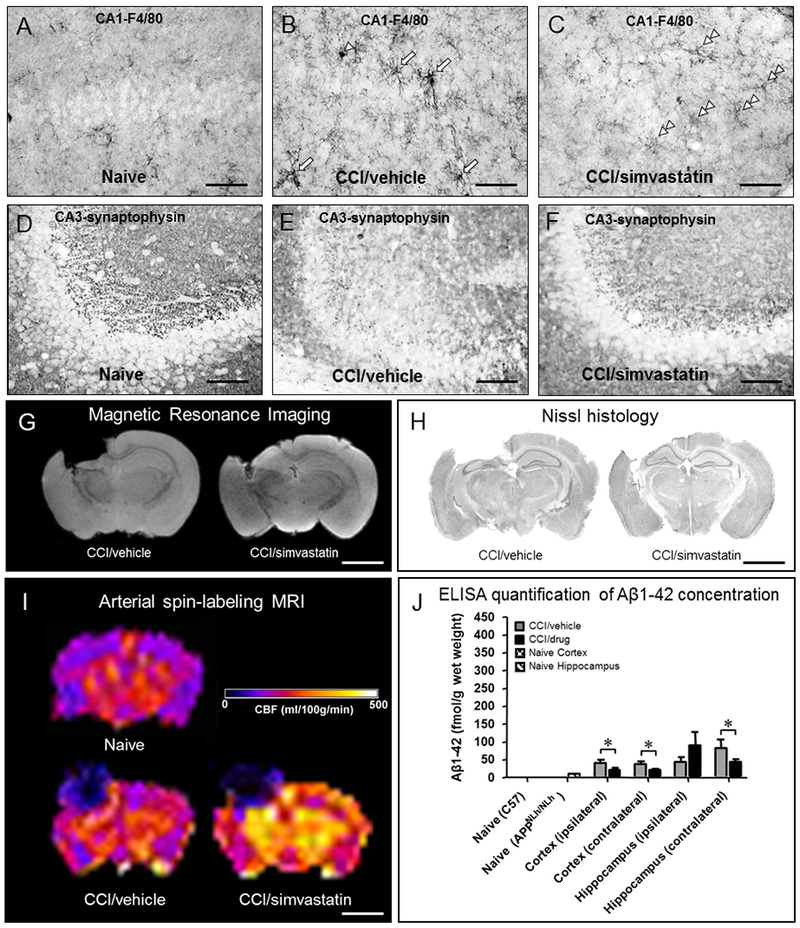
- Abrahamson EE, Ikonomovic MD, Dixon CE, DeKosky ST, 2009. Simvastatin therapy prevents brain trauma-induced increases in beta-amyloid peptide levels. Ann Neurol 66, 407–414. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
