

Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls
- PMID: 29933573
- PMCID: PMC6161106
- DOI: 10.3390/proteomes6030029
Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls
Abstract
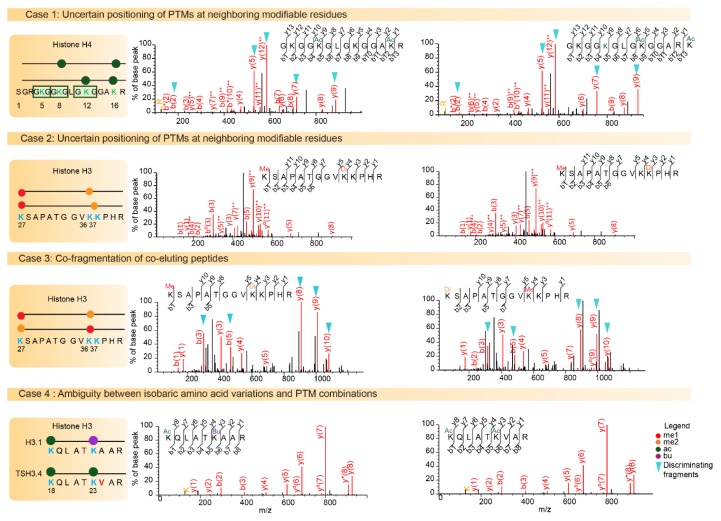
Epigenetic modifications contribute to the determination of cell fate and differentiation. The molecular mechanisms underlying histone variants and post-translational modifications (PTMs) have been studied in the contexts of development, differentiation, and disease. Antibody-based assays have classically been used to target PTMs, but these approaches fail to reveal combinatorial patterns of modifications. In addition, some histone variants are so similar to canonical histones that antibodies have difficulty distinguishing between these isoforms. Mass spectrometry (MS) has progressively developed as a powerful technology for the study of histone variants and their PTMs. Indeed, MS analyses highlighted exquisitely complex combinations of PTMs, suggesting “crosstalk” between them, and also revealed that PTM patterns are often variant-specific. Even though the sensitivity and acquisition speed of MS instruments have considerably increased alongside the development of computational tools for the study of multiple PTMs, it remains challenging to correctly describe the landscape of histone PTMs, and in particular to confidently assign modifications to specific amino acids. Here, we provide an inventory of MS-based strategies and of the pitfalls inherent to histone PTM and variant characterization, while stressing the complex interplay between PTMs and histone sequence variations. We will particularly illustrate the roles played by MS-based analyses in identifying and quantifying histone variants and modifications.
Keywords: bottom-up analysis; computational tools; crosstalk; histone variants; mass spectrometry; post-translational modifications.
Conflict of interest statement
None of the authors have any conflict of interest to declare.
Figures




References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
