

Mechanism of APTX nicked DNA sensing and pleiotropic inactivation in neurodegenerative disease
- PMID: 29934293
- PMCID: PMC6043908
- DOI: 10.15252/embj.201798875
Mechanism of APTX nicked DNA sensing and pleiotropic inactivation in neurodegenerative disease
Abstract
The failure of DNA ligases to complete their catalytic reactions generates cytotoxic adenylated DNA strand breaks. The APTX RNA-DNA deadenylase protects genome integrity and corrects abortive DNA ligation arising during ribonucleotide excision repair and base excision DNA repair, and APTX human mutations cause the neurodegenerative disorder ataxia with oculomotor ataxia 1 (AOA1). How APTX senses cognate DNA nicks and is inactivated in AOA1 remains incompletely defined. Here, we report X-ray structures of APTX engaging nicked RNA-DNA substrates that provide direct evidence for a wedge-pivot-cut strategy for 5'-AMP resolution shared with the alternate 5'-AMP processing enzymes POLβ and FEN1. Our results uncover a DNA-induced fit mechanism regulating APTX active site loop conformations and assembly of a catalytically competent active center. Further, based on comprehensive biochemical, X-ray and solution NMR results, we define a complex hierarchy for the differential impacts of the AOA1 mutational spectrum on APTX structure and activity. Sixteen AOA1 variants impact APTX protein stability, one mutation directly alters deadenylation reaction chemistry, and a dominant AOA1 variant unexpectedly allosterically modulates APTX active site conformations.
Keywords: APTX; Ataxia Oculomotor Apraxia 1; DNA repair; X‐ray crystallography; missense mutation.
© 2018 The Authors. Published under the terms of the CC BY 4.0 license.
Figures

Schematic presentation of APTX 2‐step deadenylation mechanism.
Concatemerized nicked RNA‐DNA duplex substrate bound in the structure of hAPTX‐nicked‐RNA‐DNA complex. The 6 bp upstream region is shown in blue, 8 bp downstream region, and the complementary template strand in black, 5′ ribonucleotide positioned at the nick junction in yellow, and N‐terminal α1‐helix in orange.
Transparent surface representation of two APTX protomers (Pro1, cyan and Pro2, gray) bound to a single RNA‐DNA molecule containing two nick sub‐sites in each asymmetric unit. Simulated annealing omit 2Fo‐Fc electron density for the bound nicked RNA‐DNA (magenta) illustrates APTX engagement to DNA nick induces large‐scale DNA distortions at the nick.
X‐ray structure of hAPTX‐nicked‐RNA‐DNA complex. N‐terminal α1‐helix is colored orange, HIT domain in light blue, Znf domain in light green, DNA in pink, RNA in yellow, AMP in teal, and HIT‐loop in dark green.

Molecular details of APTX‐DNA nick interface illustrate the N‐terminal α1‐helix wedges into the DNA base stack. His166 and Trp167 stack against the bases at the nick, bending the DNA duplex at a ˜90° and unwinding both the 3′ and 5′ ends of the damaged strand.
Limited chymotryptic proteolysis of APTX. The unliganded full‐length APTX was proteolysed to C1 fragment (blue) mapped to the N‐terminus of the HIT domain. The C1 fragment was further degraded to C2 (magenta) and C3 (green), and mapped to the N‐term α1‐helix (orange box). Addition of blunt‐ended DNA, nicked DNA, and transition mimic DNA (adenosine (Ade) and orthovanadate ()) substrates resulted in proteolytic protection of C1. APTX was not protected from proteolysis when incubated with relaxed circular plasmid DNA. AOA1 K197Q active site mutant that is severely defective in DNA binding displays impaired DNA‐end‐dependent proteolytic protection.
Locations of [methyl‐13C]‐labeled methionine residues (cyan) mapped in the catalytic core of APTX.
Overlays of assigned 1H‐13C HSQC spectra of [methyl‐13C]methionine‐labeled wild‐type APTX in unliganded state (black), blunt‐ended RNA‐DNA substrate bound state (cyan), and nicked RNA‐DNA substrate bound state (red). The arrows indicate resonance shifts in response to DNA substrates.
Overlays of assigned 1H‐13C HSQC spectra of [methyl‐13C]methionine‐labeled wild‐type APTX in unliganded state (black), adenosine‐VO3‐RNA‐DNA transition mimic state (blue), and nicked AMP and RNA‐DNA product bound state (purple). The arrows indicate resonance shifts in response to RNA‐DNA substrates.
Kinetic parameters of APTX processing of 5′‐adenylated RNA‐DNA substrates, nicked (red) and blunt‐ended (cyan). Mean ± 1 SD (n = 3 technical replicates) is displayed.

APTX mutations in AOA1 map to the catalytic core of APTX comprising the HIT (blue) and Znf (green) domains.
Solubility of 17 AOA1‐linked APTX missense and nonsense mutants expressed in E. coli was assessed by Western blotting. With the exception of two nonsense variants (R247X and W279X), all proteins expressed at comparable levels. The soluble protein fractions revealed marked variability in solubility.
Thermal melting profile of APTX mutants. Unfolding of APTX proteins was monitored using SYPRO Orange fluorescent dye. The midpoint temperature (T m) for APTX wild‐type and mutants in unliganded (bottom) and AMP bound (top) conditions is displayed. X = unable to determine T m, *P‐value < 0.05, **P‐value < 0.01, N.S. = not‐significant, two‐tailed t‐test. Mean ± 1 SD (three technical replicates) is shown.
Deadenylation activity of APTX mutants. Tenfold dilutions of APTX mutant proteins were tested for deadenylation on a TAMRA labeled 5′‐adenylated nicked RNA‐DNA substrate at 25 and 37°C. Reaction products were resolved on 15% TBE‐urea denaturing gels and visualized by fluorescence scan. Fold increase in protein to reach 50% activity relative to wild‐type APTX is displayed. Mean ± 1 SD (3 technical replicates) is displayed.
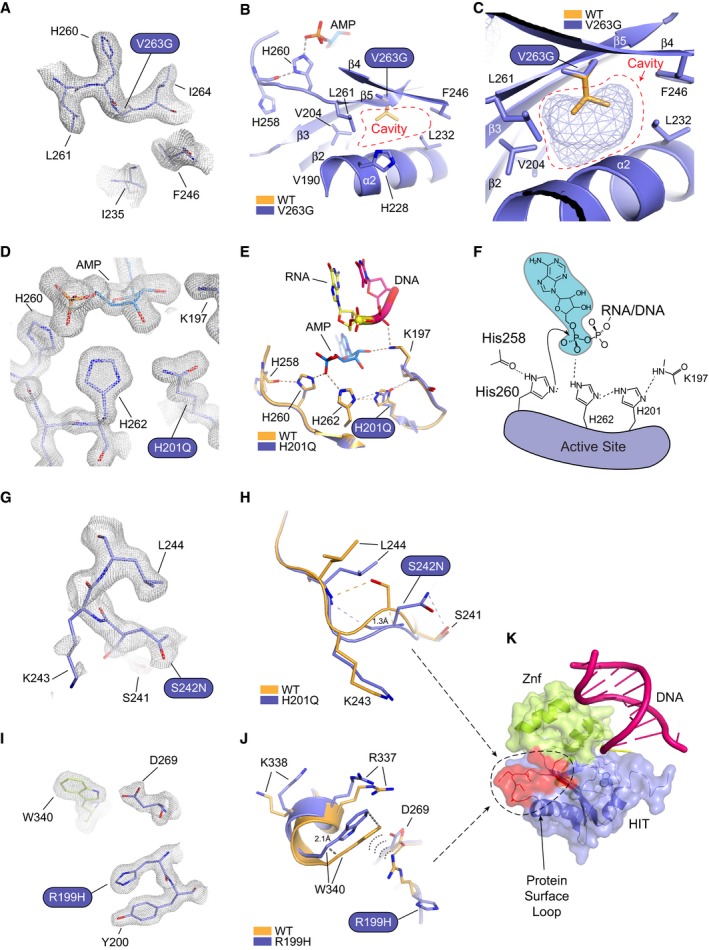
Omit 2Fo‐Fc electron density for the V263G mutant.
X‐ray structure of the V263G mutant (blue) overlaid upon wild‐type APTX (orange). Val263 maps to β5 and is a hydrophobic core residue engaging in close Van der Waals interactions with neighboring hydrophobic residues. The structure of V263G shows the mutation creates a large cavity in the hydrophobic core, resulted from the loss of the valine side chain.
Mesh space filled presentation illustrates a cavity in APTX hydrophobic core created by the V263G mutation.
Omit 2Fo‐Fc electron density for the H201Q mutant.
X‐ray structure of the H201Q mutant (blue) overlaid upon wild‐type APTX (orange). His201 participates in a charge relay mechanism that stabilizes the transition state of the APTX deadenylation reaction.
Schematic presentation of the APTX active site. A charge relay consists of the main‐chain amide of Lys197, His201, and His262 that hydrogen bonds to the scissile phosphate of the 5′‐adenylate phosphoanhydride. The structure of H201Q reveals the disruption to the charge relay network.
Omit 2Fo‐Fc electron density for the S242N mutant.
X‐ray structure of the S242N mutant (blue) overlaid upon wild‐type APTX (orange). Ser242 maps to a surface loop. The native loop conformation in wild‐type APTX is stabilized through hydrogen bond between Ser242 hydroxyl and Leu244 main‐chain amide. The serine to asparagine mutation causes rearrangements of side‐chain contacts resulting in an altered loop structure with a 1.3 Å shift at the Ser242 Cα.
Omit 2Fo‐Fc electron density for the R199H mutant.
X‐ray structure of the R199H mutant (blue) overlaid upon wild‐type APTX (orange). Arg199 forms a salt bridge with Asp269 and a cation‐π interaction with C‐terminal residue Trp340. The arginine to histidine mutation causes the loss of these contacts resulting in an altered protein surface structure with a 2.1 Å shift from the protein core.
Surface representation of APTX RNA‐DNA complex showing both S242N and R199H mapped on a putative protein binding surface.
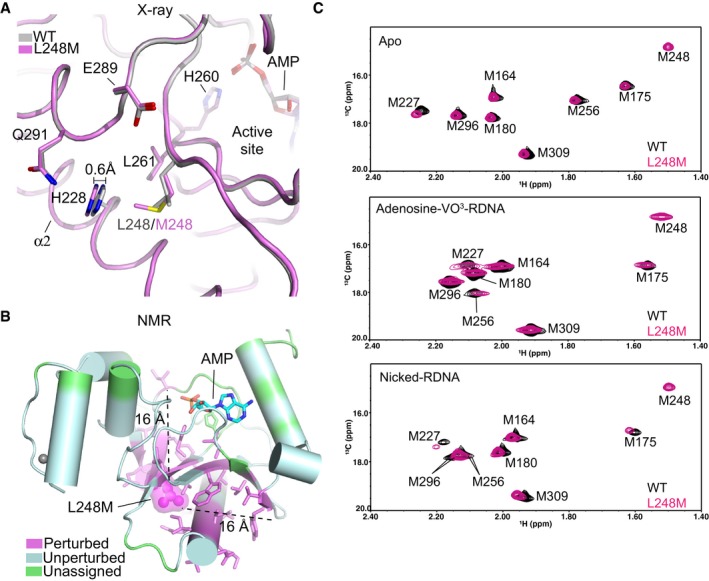
X‐ray structural overlays of AOA1 mutant L248M (purple) and wild‐type APTX (gray).
Location of residues exhibiting 1H‐15N NMR chemical shift perturbation (also see Appendix Fig S8) due to the L248M mutation. Perturbed (magenta), unperturbed (cyan), unassigned (green).
Assigned 1H‐13C HSQC spectral overlay of [methyl‐13C]methionine‐labeled wild‐type APTX and L248M mutant in APO (top), transition state (middle), and product bound state (bottom).

The APTX α1 wedges into the damaged nick, inducing a ˜90° bend in the DNA and exposing both the 3′ and 5′ termini. In addition to APTX, DNA polymerase β (POLβ) and FEN1 nucleases provide alternate processing pathways for 5′‐AMP repair.
Direct reversal of adenylation by APTX.
Endonucleolytic incision of the damaged 5′ adenylated strand by FEN1.
Removal of adenylated 5′‐deoxyribose phosphate (5′‐dRP) by POLβ.

References
-
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner RD, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2011) The Phenix software for automated determination of macromolecular structures. Methods 55: 94–106 - PMC - PubMed
-
- Ahel I, Rass U, El‐Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC (2006) The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 443: 713–716 - PubMed
-
- Baba Y, Uitti RJ, Boylan KB, Uehara Y, Yamada T, Farrer MJ, Couchon E, Batish SD, Wszolek ZK (2007) Aprataxin (APTX) gene mutations resembling multiple system atrophy. Parkinsonism Relat Disord 13: 139–142 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
