

The ASD Living Biology: from cell proliferation to clinical phenotype
- PMID: 29934544
- PMCID: PMC6309606
- DOI: 10.1038/s41380-018-0056-y
The ASD Living Biology: from cell proliferation to clinical phenotype
Abstract
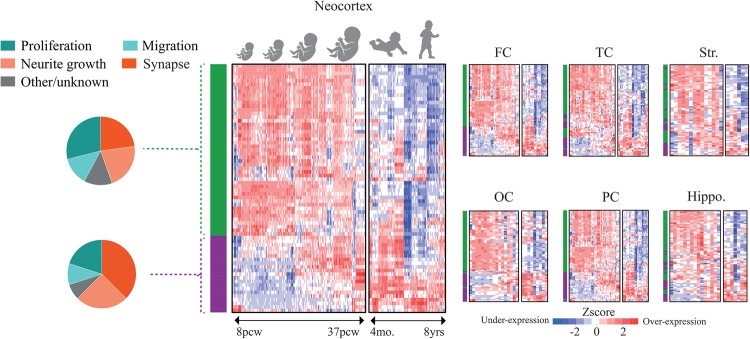
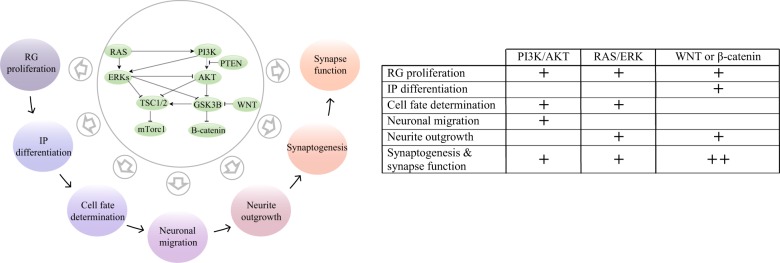
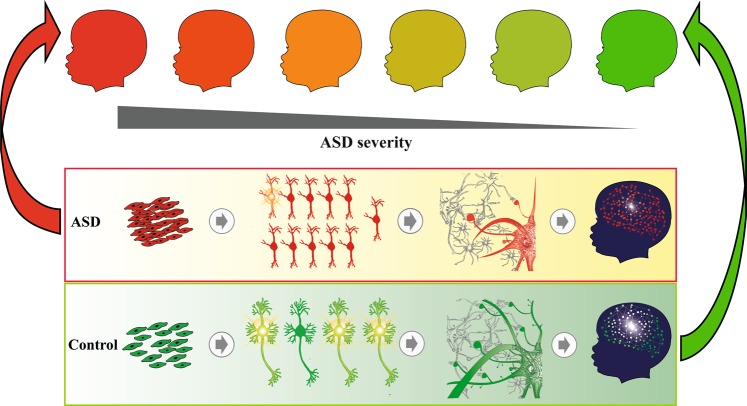
Autism spectrum disorder (ASD) has captured the attention of scientists, clinicians and the lay public because of its uncertain origins and striking and unexplained clinical heterogeneity. Here we review genetic, genomic, cellular, postmortem, animal model, and cell model evidence that shows ASD begins in the womb. This evidence leads to a new theory that ASD is a multistage, progressive disorder of brain development, spanning nearly all of prenatal life. ASD can begin as early as the 1st and 2nd trimester with disruption of cell proliferation and differentiation. It continues with disruption of neural migration, laminar disorganization, altered neuron maturation and neurite outgrowth, disruption of synaptogenesis and reduced neural network functioning. Among the most commonly reported high-confidence ASD (hcASD) genes, 94% express during prenatal life and affect these fetal processes in neocortex, amygdala, hippocampus, striatum and cerebellum. A majority of hcASD genes are pleiotropic, and affect proliferation/differentiation and/or synapse development. Proliferation and subsequent fetal stages can also be disrupted by maternal immune activation in the 1st trimester. Commonly implicated pathways, PI3K/AKT and RAS/ERK, are also pleiotropic and affect multiple fetal processes from proliferation through synapse and neural functional development. In different ASD individuals, variation in how and when these pleiotropic pathways are dysregulated, will lead to different, even opposing effects, producing prenatal as well as later neural and clinical heterogeneity. Thus, the pathogenesis of ASD is not set at one point in time and does not reside in one process, but rather is a cascade of prenatal pathogenic processes in the vast majority of ASD toddlers. Despite this new knowledge and theory that ASD biology begins in the womb, current research methods have not provided individualized information: What are the fetal processes and early-age molecular and cellular differences that underlie ASD in each individual child? Without such individualized knowledge, rapid advances in biological-based diagnostic, prognostic, and precision medicine treatments cannot occur. Missing, therefore, is what we call ASD Living Biology. This is a conceptual and paradigm shift towards a focus on the abnormal prenatal processes underlying ASD within each living individual. The concept emphasizes the specific need for foundational knowledge of a living child's development from abnormal prenatal beginnings to early clinical stages. The ASD Living Biology paradigm seeks this knowledge by linking genetic and in vitro prenatal molecular, cellular and neural measurements with in vivo post-natal molecular, neural and clinical presentation and progression in each ASD child. We review the first such study, which confirms the multistage fetal nature of ASD and provides the first in vitro fetal-stage explanation for in vivo early brain overgrowth. Within-child ASD Living Biology is a novel research concept we coin here that advocates the integration of in vitro prenatal and in vivo early post-natal information to generate individualized and group-level explanations, clinically useful prognoses, and precision medicine approaches that are truly beneficial for the individual infant and toddler with ASD.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures





References
-
- Kanner L. Autistic disturbances of affective contact. Nerv Child. 1943;2:217–50. - PubMed
-
- Constantino JN, Marrus N. The early origins of autism. Child Adolesc Psychiatr Clin N Am. 2017;26:555–70. - PubMed
-
- Dawson G, Meltzoff AN, Osterling J, Rinaldi J, Brown E. Children with autism fail to orient to naturally occurring social stimuli. J Autism Dev Disord. 1998;28:479–85. - PubMed
-
- Courchesne E, Karns C, Davis HR, Ziccardi R, Carper R, Tigue Z, et al. Unusual brain growth patterns in early life in patients with autistic disorder: An MRI study. Neurology. 2001;57:245–54. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
