

High-Throughput Functional Analysis Distinguishes Pathogenic, Nonpathogenic, and Compensatory Transcriptional Changes in Neurodegeneration
- PMID: 29936182
- PMCID: PMC6082401
- DOI: 10.1016/j.cels.2018.05.010
High-Throughput Functional Analysis Distinguishes Pathogenic, Nonpathogenic, and Compensatory Transcriptional Changes in Neurodegeneration
Abstract
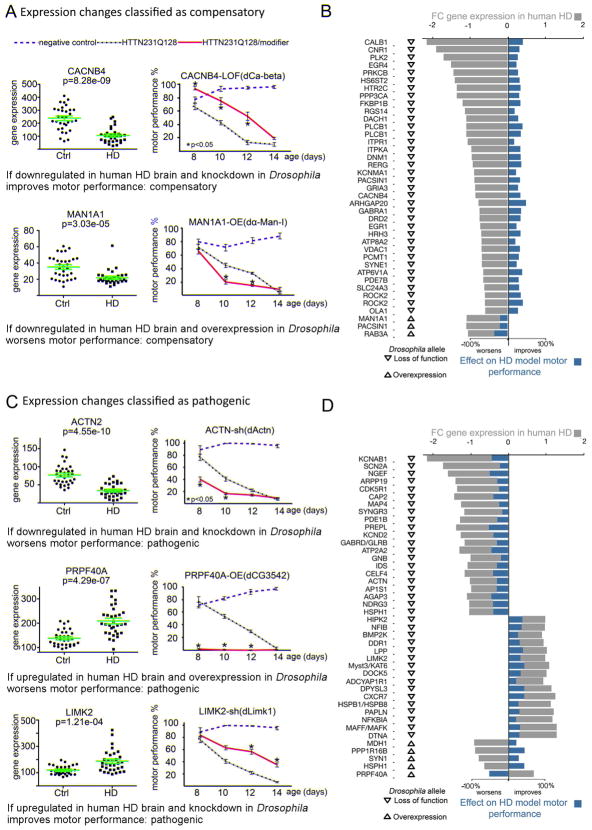
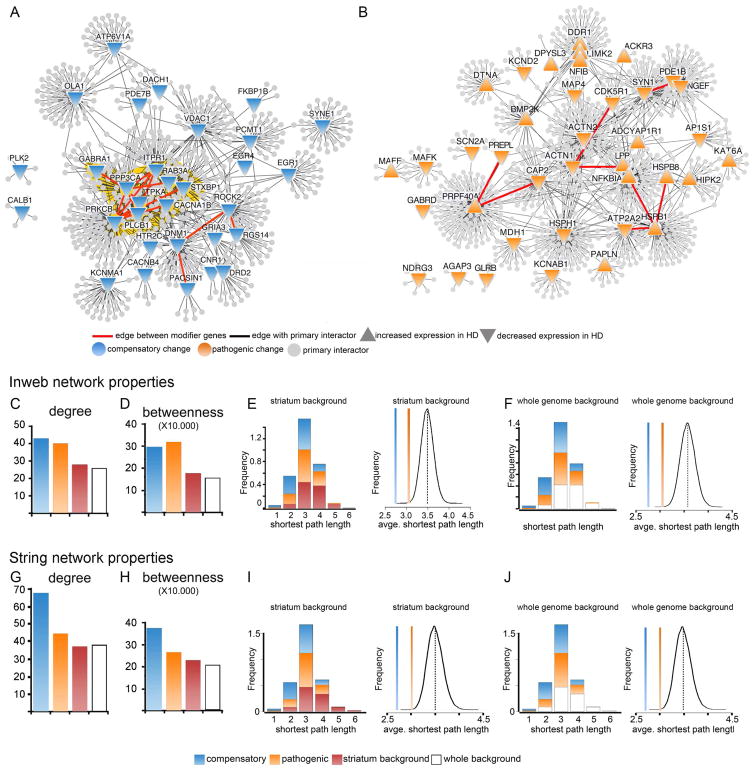
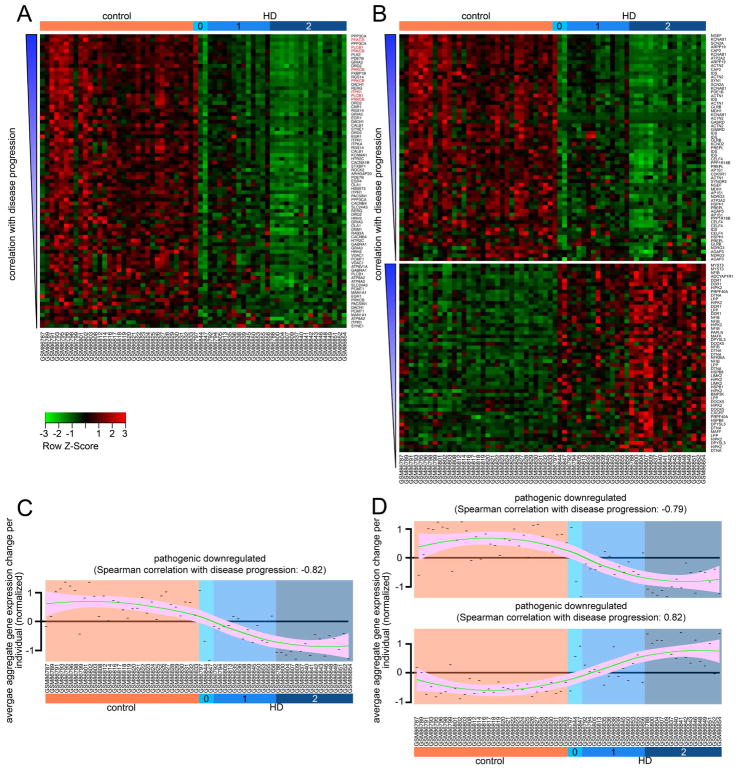
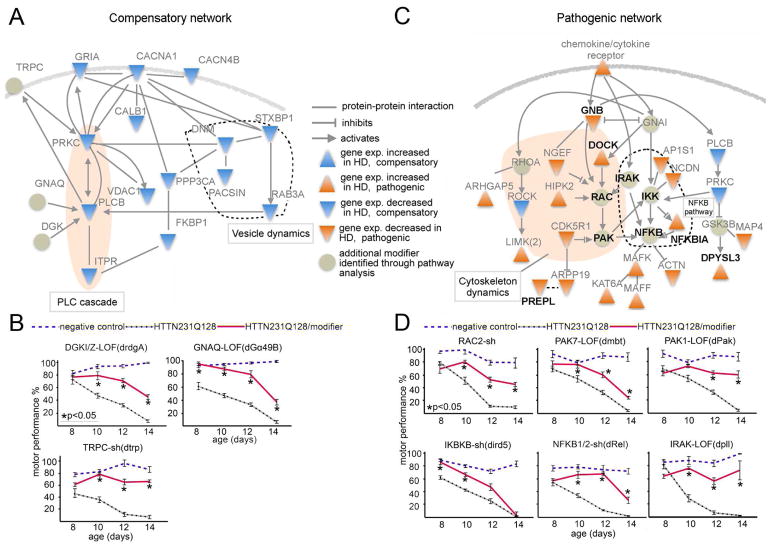
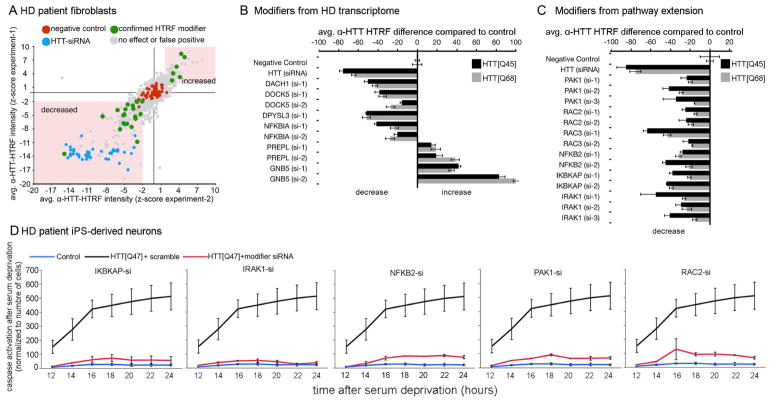
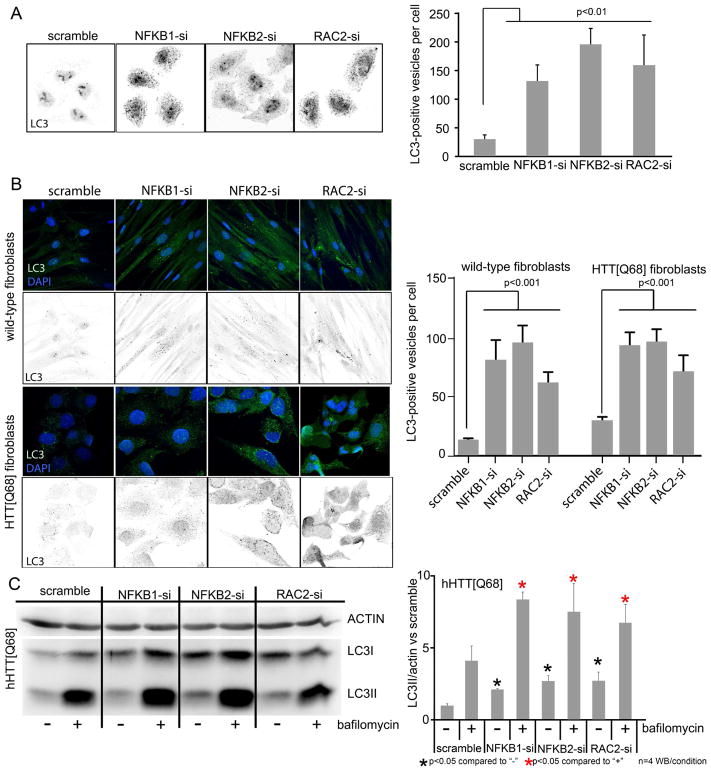
Discriminating transcriptional changes that drive disease pathogenesis from nonpathogenic and compensatory responses is a daunting challenge. This is particularly true for neurodegenerative diseases, which affect the expression of thousands of genes in different brain regions at different disease stages. Here we integrate functional testing and network approaches to analyze previously reported transcriptional alterations in the brains of Huntington disease (HD) patients. We selected 312 genes whose expression is dysregulated both in HD patients and in HD mice and then replicated and/or antagonized each alteration in a Drosophila HD model. High-throughput behavioral testing in this model and controls revealed that transcriptional changes in synaptic biology and calcium signaling are compensatory, whereas alterations involving the actin cytoskeleton and inflammation drive disease. Knockdown of disease-driving genes in HD patient-derived cells lowered mutant Huntingtin levels and activated macroautophagy, suggesting a mechanism for mitigating pathogenesis. Our multilayered approach can thus untangle the wealth of information generated by transcriptomics and identify early therapeutic intervention points.
Keywords: Huntington disease; NFKB; RAC2; actin cytoskeleton; autophagy; calcium signaling; compensatory changes; inflammation; synaptic biology; transcriptome.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Ament SA, Pearl JR, Grindeland A, St Claire J, Earls JC, Kovalenko M, Gillis T, Mysore J, Gusella JF, Lee JM, et al. High resolution time-course mapping of early transcriptom-ic, molecular and cellular phenotypes in Huntington’s disease CAG knock-in mice across multiple genetic backgrounds. Hum Mol Genet. 2017;26:913–922. - PMC - PubMed
-
- Andre R, Carty L, Tabrizi SJ. Disruption of immune cell function by mutant huntingtin in Huntington’s disease pathogenesis. Curr Opin Pharmacol. 2016;26:33–38. - PubMed
-
- Branco J, Al-Ramahi I, Ukani L, Perez AM, Fernandez-Funez P, Rincon-Limas D, Botas J. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum Mol Genet. 2008;17:376–390. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
