

Analytical Validation of a Hybrid Capture-Based Next-Generation Sequencing Clinical Assay for Genomic Profiling of Cell-Free Circulating Tumor DNA
- PMID: 29936259
- PMCID: PMC6593250
- DOI: 10.1016/j.jmoldx.2018.05.004
Analytical Validation of a Hybrid Capture-Based Next-Generation Sequencing Clinical Assay for Genomic Profiling of Cell-Free Circulating Tumor DNA
Abstract
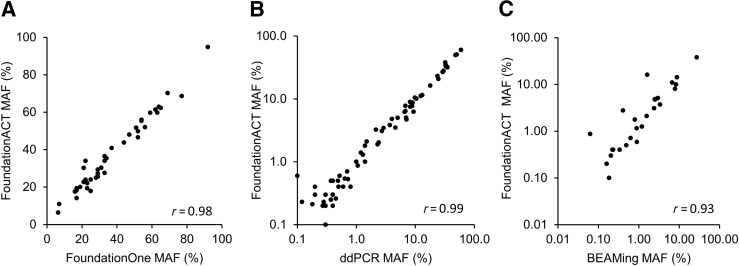
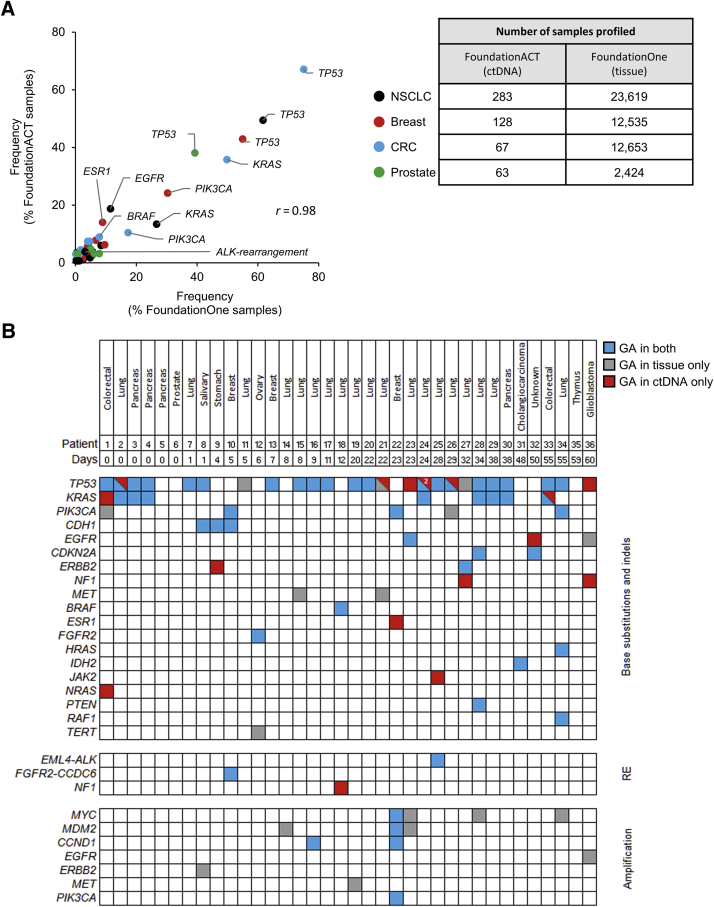
Genomic profiling of circulating tumor DNA derived from cell-free DNA (cfDNA) in blood can provide a noninvasive method for detecting genomic biomarkers to guide clinical decision making for cancer patients. We developed a hybrid capture-based next-generation sequencing assay for genomic profiling of circulating tumor DNA from blood (FoundationACT). High-sequencing coverage and molecular barcode-based error detection enabled accurate detection of genomic alterations, including short variants (base substitutions, short insertions/deletions) and genomic re-arrangements at low allele frequencies (AFs), and copy number amplifications. Analytical validation was performed on 2666 reference alterations. The assay achieved >99% overall sensitivity (95% CI, 99.1%-99.4%) for short variants at AF >0.5%, >95% sensitivity (95% CI, 94.2%-95.7%) for AF 0.25% to 0.5%, and 70% sensitivity (95% CI, 68.2%-71.5%) for AF 0.125% to 0.25%. No false positives were detected in 62 samples from healthy volunteers. Genomic alterations detected by FoundationACT demonstrated high concordance with orthogonal assays run on the same clinical cfDNA samples. In 860 routine clinical FoundationACT cases, genomic alterations were detected in cfDNA at comparable frequencies to tissue; for the subset of cases with temporally matched tissue and blood samples, 75% of genomic alterations and 83% of short variant mutations detected in tissue were also detected in cfDNA. On the basis of analytical validation results, FoundationACT has been approved for use in our Clinical Laboratory Improvement Amendments-certified/College of American Pathologists-accredited/New York State-approved laboratory.
Copyright © 2018 American Society for Investigative Pathology and the Association for Molecular Pathology. Published by Elsevier Inc. All rights reserved.
Figures





References
-
- Hartmaier R.J., Albacker L., Chmielecki J., Bailey M., He J., Goldberg M.E., Ramkissoon S., Suh J., Elvin J.A., Chiacchia S., Frampton G.M., Ross J.S., Miller V., Stephens P.J., Lipson D. High-throughput genomic profiling of adult solid tumors reveals novel insights into cancer pathogenesis. Cancer Res. 2017;77:2464–2475. - PubMed
-
- Eisenberg R., Varmus H. Insurance for broad genomic tests in oncology. Science. 2017;358:1133–1134. - PubMed
-
- Swisher E.M., Lin K.K., Oza A.M., Scott C.L., Giordano H., Sun J., Konecny G.E., Coleman R.L., Tinker A.V., O'Malley D.M., Kristeleit R.S., Ma L., Bell-McGuinn K.M., Brenton J.D., Cragun J.M., Oaknin A., Ray-Coquard I., Harrell M.I., Mann E., Kaufmann S.H., Floquet A., Leary A., Harding T.C., Goble S., Maloney L., Isaacson J., Allen A.R., Rolfe L., Yelensky R., Raponi M., McNeish I.A. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
