

Biological classification with RNA-seq data: Can alternatively spliced transcript expression enhance machine learning classifiers?
- PMID: 29941426
- PMCID: PMC6097660
- DOI: 10.1261/rna.062802.117
Biological classification with RNA-seq data: Can alternatively spliced transcript expression enhance machine learning classifiers?
Abstract
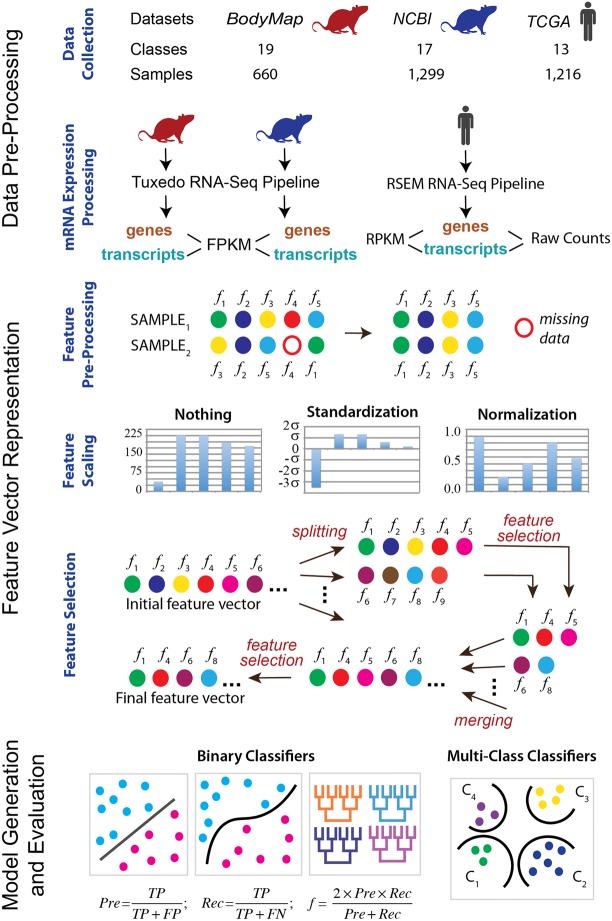
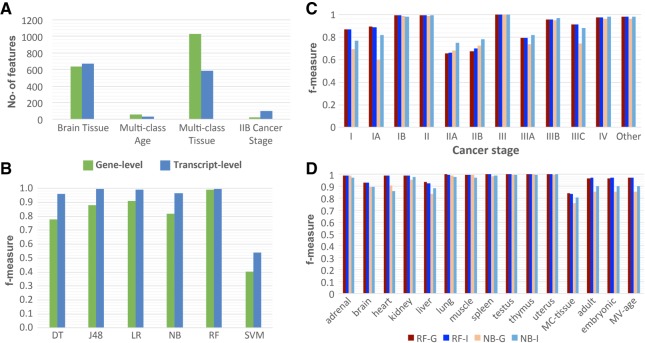
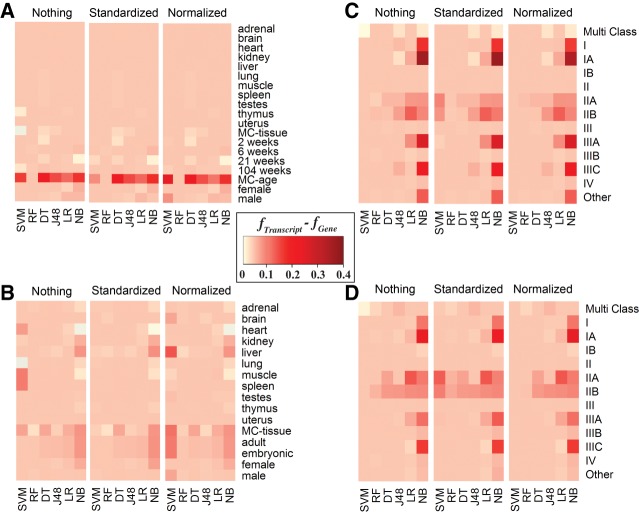
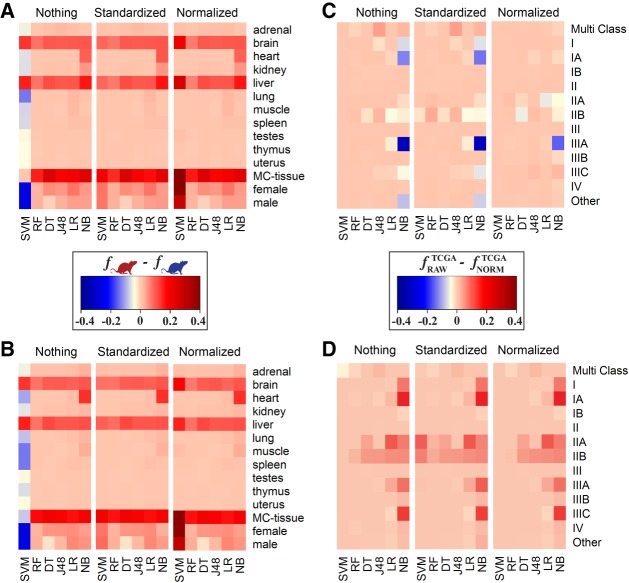
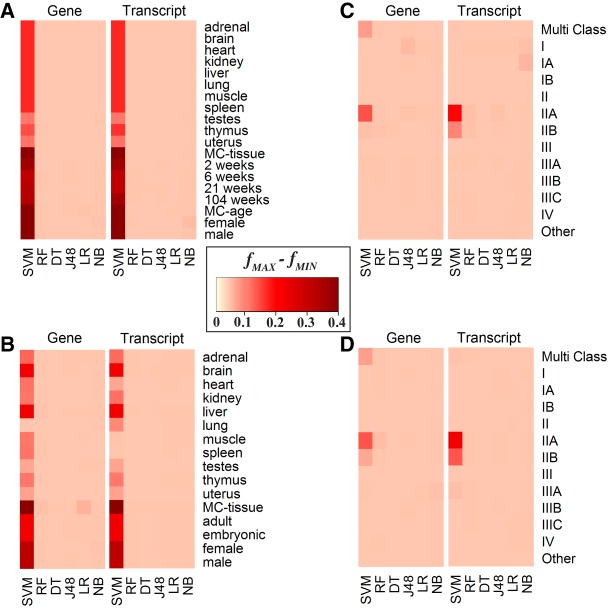
RNA sequencing (RNA-seq) is becoming a prevalent approach to quantify gene expression and is expected to gain better insights into a number of biological and biomedical questions compared to DNA microarrays. Most importantly, RNA-seq allows us to quantify expression at the gene or transcript levels. However, leveraging the RNA-seq data requires development of new data mining and analytics methods. Supervised learning methods are commonly used approaches for biological data analysis that have recently gained attention for their applications to RNA-seq data. Here, we assess the utility of supervised learning methods trained on RNA-seq data for a diverse range of biological classification tasks. We hypothesize that the transcript-level expression data are more informative for biological classification tasks than the gene-level expression data. Our large-scale assessment utilizes multiple data sets, organisms, lab groups, and RNA-seq analysis pipelines. Overall, we performed and assessed 61 biological classification problems that leverage three independent RNA-seq data sets and include over 2000 samples that come from multiple organisms, lab groups, and RNA-seq analyses. These 61 problems include predictions of the tissue type, sex, or age of the sample, healthy or cancerous phenotypes, and pathological tumor stages for the samples from the cancerous tissue. For each problem, the performance of three normalization techniques and six machine learning classifiers was explored. We find that for every single classification problem, the transcript-based classifiers outperform or are comparable with gene expression-based methods. The top-performing techniques reached a near perfect classification accuracy, demonstrating the utility of supervised learning for RNA-seq based data analysis.
Keywords: RNA-seq; alternative splicing; classification; gene expression; machine learning.
© 2018 Johnson et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures
References
-
- Achim K, Pettit JB, Saraiva LR, Gavriouchkina D, Larsson T, Arendt D, Marioni JC. 2015. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat Biotechnol 33: 503–509. - PubMed
-
- Asur S, Raman P, Otey ME, Parthasarathy S. 2006. A model-based approach for mining membrane protein crystallization trials. Bioinformatics 22: e40–e48. - PubMed
-
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338: 1587–1593. - PubMed
-
- Boulesteix AL, Janitza S, Kruppa J, König IR. 2012. Overview of random forest methodology and practical guidance with emphasis on computational biology and bioinformatics. Wiley Interdiscip Rev Data Min Knowl Discov 2: 493–507.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous