

Identification of a Novel Homozygous Splice-Site Mutation in SCARB2 that Causes Progressive Myoclonus Epilepsy with or without Renal Failure
- PMID: 29941711
- PMCID: PMC6032684
- DOI: 10.4103/0366-6999.235113
Identification of a Novel Homozygous Splice-Site Mutation in SCARB2 that Causes Progressive Myoclonus Epilepsy with or without Renal Failure
Abstract
Background: Progressive myoclonus epilepsies (PMEs) comprise a group of rare genetic disorders characterized by action myoclonus, epileptic seizures, and ataxia with progressive neurologic decline. Due to clinical and genetic heterogeneity of PMEs, it is difficult to decide which genes are affected. The aim of this study was to report an action myoclonus with or without renal failure syndrome (EPM4) family and summarize the clinical and genetic characteristics of all reported EPM4 patients.
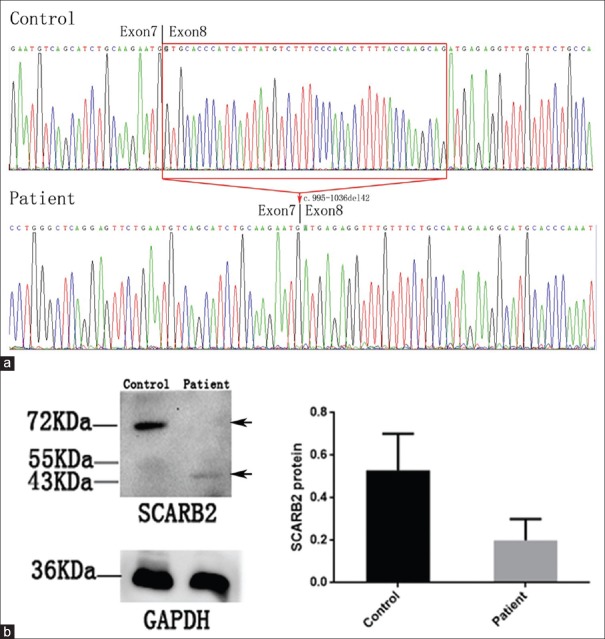
Methods: In the present study, targeted next-generation sequencing (NGS) was applied to screen causative genes in a Chinese PME family. The candidate variant was further confirmed by cosegregation analysis and further functional analysis, including the reverse transcription polymerase chain reaction and Western blot of the proband's muscle. Moreover, literature data on the clinical and mutational features of all reported EPM4 patients were reviewed.
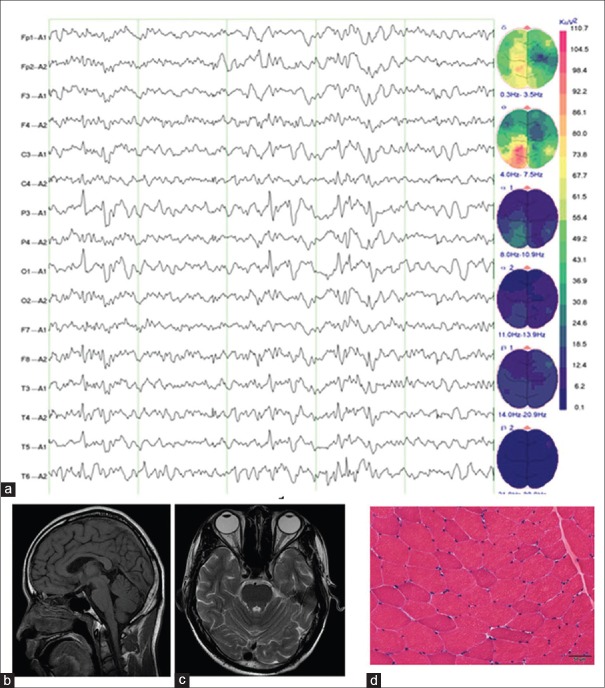
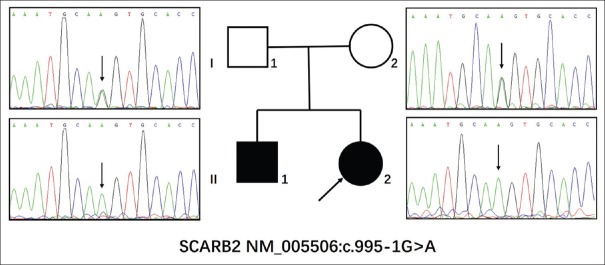
Results: The gene analysis revealed a novel homozygous splicing mutation (c.995-1G>A) of the SCARB2 gene in two brothers. Further functional analysis revealed that this mutation led to loss function of the SCARB2 protein. The classification of the candidate variant, according to the American College of Medical Genetics and Genomics standards and guidelines and functional analysis, was pathogenic. Therefore, these two brothers were finally diagnostically confirmed as EPM4.
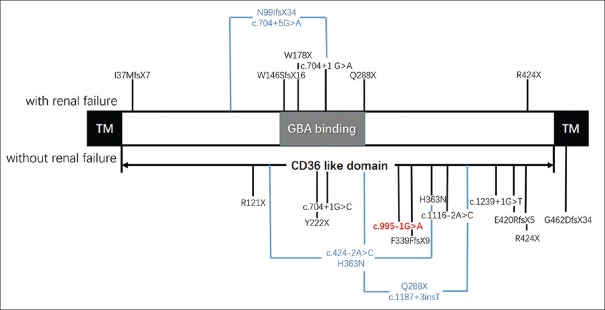
Conclusions: These present results suggest the potential for targeted NGS to conduct a more rapid and precise diagnosis for PME patients. A literature review revealed that mutations in the different functional domains of SCARB2 appear to be associated with the phenotype of EPM4.
SCARB2基因新剪接位点纯合突变导致进行性肌阵挛癫痫伴或不伴肾功能衰竭摘要背景:进行性癫痫性肌阵挛(PMEs)是一组以肌阵挛、癫痫发作、共济失调及神经系统功能进行性减退为特点的罕见病。由于该病具有临床和基因异质性,明确疾病的致病基因有一定难度。 本文对一例EPM4家系进行了报道并总结了所有已报道EMP4患者的临床与基因突变特点。 方法:本研究应用目标区域测序在一个家系中检测 PMEs致病基因并对候选基因变异位点进行家系共分离分析以及对先证者肌肉细胞进行基因表达水平和蛋白表达水平分析。同时,回顾并总结所有已报道EPM4临床和突变特点的文献。 结果:通过基因分析发现,先证者及其兄长均携带SCARB2基因c.995-1G>A剪切位点突变。进一步实验发现该突变导致SCARB2蛋白功能缺失。根据美国医学遗传学和基因组学学院(ACMG)标准和指南,该候选变异位点为致病突变,该兄弟最终被确诊EPM4。 结论:靶向二代测序未来有望成为更加快速精准诊断PMEs的方法。通过回顾相关文献表明EPM4的表型与突变位点所在SCARB2基因功能域有关。.
Keywords: Gene; Progressive Myoclonus Epilepsies; Progressive Myoclonus Epilepsy with or without Renal Failure; SCARB2; Targeted Next-Generation Sequencing.
Conflict of interest statement
There are no conflicts of interest
Figures
Similar articles
-
A novel homozygous splice-site mutation in SCARB2 is associated with progressive myoclonic epilepsy with renal failure.Neurol Sci. 2021 Dec;42(12):5077-5085. doi: 10.1007/s10072-021-05196-0. Epub 2021 Mar 26. Neurol Sci. 2021. PMID: 33772352
-
Progressive myoclonus epilepsy without renal failure in a Chinese family with a novel mutation in SCARB2 gene and literature review.Seizure. 2018 Apr;57:80-86. doi: 10.1016/j.seizure.2018.03.015. Epub 2018 Mar 14. Seizure. 2018. PMID: 29605618 Review.
-
Clinical and neurophysiologic features of progressive myoclonus epilepsy without renal failure caused by SCARB2 mutations.Epilepsia. 2011 Dec;52(12):2356-63. doi: 10.1111/j.1528-1167.2011.03307.x. Epub 2011 Nov 2. Epilepsia. 2011. PMID: 22050460
-
Novel SCARB2 mutation in action myoclonus-renal failure syndrome and evaluation of SCARB2 mutations in isolated AMRF features.BMC Neurol. 2011 Oct 27;11:134. doi: 10.1186/1471-2377-11-134. BMC Neurol. 2011. PMID: 22032306 Free PMC article.
-
SCARB2/LIMP2 deficiency in action myoclonus-renal failure syndrome.Epileptic Disord. 2016 Sep 1;18(S2):63-72. doi: 10.1684/epd.2016.0843. Epileptic Disord. 2016. PMID: 27582254 Review.
Cited by
-
Deficiency of Glucocerebrosidase Activity beyond Gaucher Disease: PSAP and LIMP-2 Dysfunctions.Int J Mol Sci. 2024 Jun 16;25(12):6615. doi: 10.3390/ijms25126615. Int J Mol Sci. 2024. PMID: 38928321 Free PMC article.
-
Myoclonus-Ataxia Syndromes: A Diagnostic Approach.Mov Disord Clin Pract. 2020 Nov 3;8(1):9-24. doi: 10.1002/mdc3.13106. eCollection 2021 Jan. Mov Disord Clin Pract. 2020. PMID: 33426154 Free PMC article. Review.
-
An elongated tract of polyQ in the carboxyl‑terminus of human α1A calcium channel induces cell apoptosis by nuclear translocation.Oncol Rep. 2020 Jul;44(1):156-164. doi: 10.3892/or.2020.7592. Epub 2020 Apr 22. Oncol Rep. 2020. PMID: 32626992 Free PMC article.
-
Genotype-Phenotype correlations of SCARB2 associated clinical presentation: a case report and in-depth literature review.BMC Neurol. 2022 Mar 28;22(1):122. doi: 10.1186/s12883-022-02628-y. BMC Neurol. 2022. PMID: 35346091 Free PMC article.
References
-
- Kälviäinen R. Progressive myoclonus epilepsies. Semin Neurol. 2015;35:293–9. doi: 10.1055/s-0035-1552620. - PubMed
-
- Pennacchio LA, Lehesjoki AE, Stone NE, Willour VL, Virtaneva K, Miao J, et al. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1) Science. 1996;271:1731–4. doi: 101126/science27152561731. - PubMed
-
- Gómez-Garre P, Sanz Y, Rodríguez De Córdoba SR, Serratosa JM. Mutational spectrum of the EPM2A gene in progressive myoclonus epilepsy of Lafora: High degree of allelic heterogeneity and prevalence of deletions. Eur J Hum Genet. 2000;8:946–54. doi: 10.1038/sj.ejhg.5200571. - PubMed
-
- Gómez-Abad C, Gómez-Garre P, Gutiérrez-Delicado E, Saygi S, Michelucci R, Tassinari CA, et al. Lafora disease due to EPM2B mutations: A clinical and genetic study. Neurology. 2005;64:982–6. doi: 10.1212/01.WNL.0000154519.10805.F7. - PubMed
-
- Berkovic SF, Mazarib A, Walid S, Neufeld MY, Manelis J, Nevo Y, et al. A new clinical and molecular form of Unverricht-Lundborg disease localized by homozygosity mapping. Brain. 2005;128:652–8. doi: 10.1093/brain/awh377. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
