

doi: 10.1038/s41592-018-0036-9.
Epub 2018 Jun 25.
DeTiN: overcoming tumor-in-normal contamination
Affiliations
- PMID: 29941871
- PMCID: PMC6528031
- DOI: 10.1038/s41592-018-0036-9
Item in Clipboard
DeTiN: overcoming tumor-in-normal contamination
Nat Methods.
2018 Jul.
Abstract
Comparison of sequencing data from a tumor sample with data from a matched germline control is a key step for accurate detection of somatic mutations. Detection sensitivity for somatic variants is greatly reduced when the matched normal sample is contaminated with tumor cells. To overcome this limitation, we developed deTiN, a method that estimates the tumor-in-normal (TiN) contamination level and, in cases affected by contamination, improves sensitivity by reclassifying initially discarded variants as somatic.
Conflict of interest statement
Competing Financial Interests Statement:
C.J.W. is a co-founder of Neon Therapeutics and a member of its scientific advisory board.
Figures
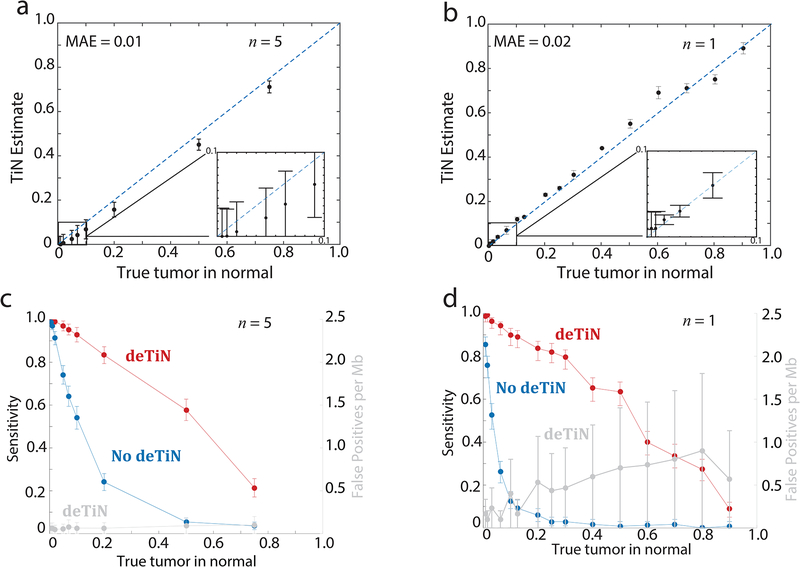
(a) TiN estimates at different in silico simulated TiN levels. (b) deTiN estimates at different in vitro mixed TiN levels. MAE = mean absolute error. (c, d) Sensitivity to detect mutations with deTiN (red) and without deTiN (blue) at (c) different in silico simulated TiN levels and (d) in vitro mixed TiN levels. (a, c) deTiN results from n=5 in silico independent simulation experiments. Dots represent weighted average and error bars represent standard errors. (b, d) Results from n=1 sequencing experiment. Error bars depict 95% confidence intervals on TiN estimates. (a, b) Dotted blue lines indicate y=x.
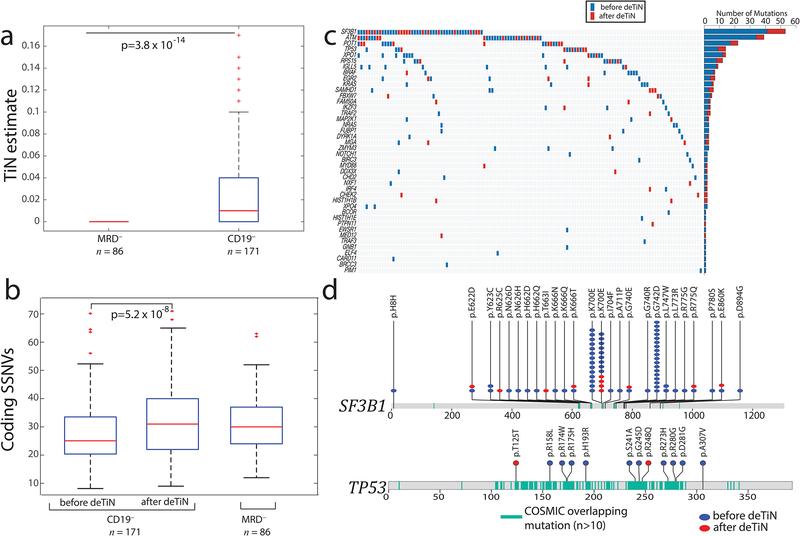
(a) TiN estimates for CD19– selected (normal) blood compared with whole blood from minimal residual disease negative (MRD–) patients. Box plot: median TiN value (red line), box represents Q1 and Q3 quartiles, whiskers represent the most extreme data points that are not outliers. Outliers are denoted with red crosses and represent data points out side the range [Q1 - 1.5 IQR, Q3 + 1.5 IQR] where IQR is the interquartile range. P value is calculated using two-tailed Mann–Whitney test (n=257 independent patient samples). (b) Mutation rate in samples pre- and post-application of deTiN stratified by normal sample type. Box plot and P value as in panel a. (c) Heat map and bar plot illustrating recovery of SSNVs in the CLL cohort. Samples are in columns, genes in rows. Blue boxes indicate variants detected prior to deTiN (“without deTiN”); red boxes indicate additional variants recovered by deTiN (“with deTiN”). (d) Stick plots showing mutation data in SF3B1 and TP53. Amino acid positions of recurrent COSMIC mutations are highlighted in teal. Blue circles indicate variants detected prior to deTiN; red circles indicate variants recovered by deTiN.
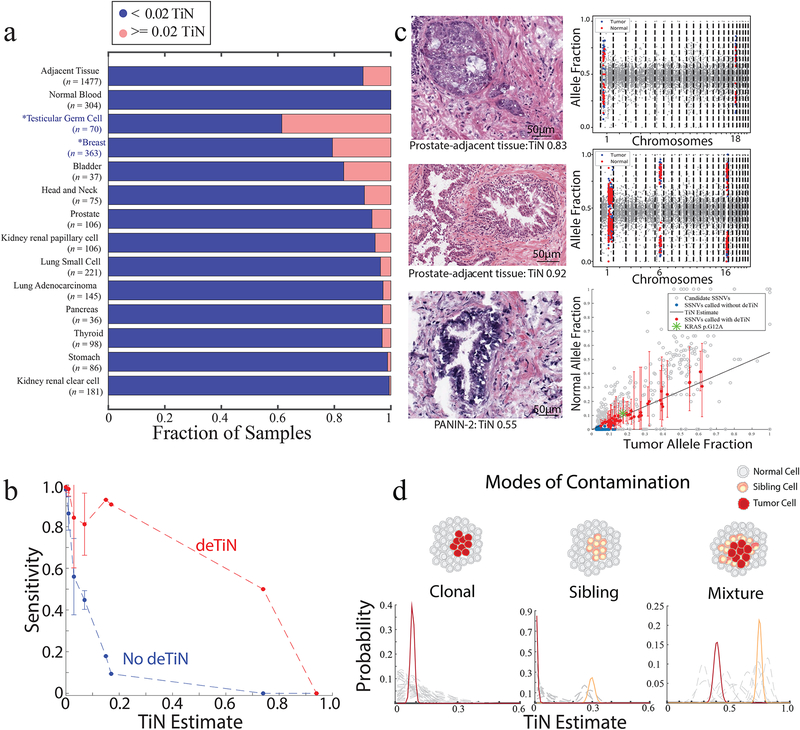
(a) Fraction of contaminated samples (pink; TiN≥0.02) when using different sources for normal tissue (tumor-adjacent normal tissue and peripheral blood) and, in cases with tumor-adjacent normal, stratified by tumor type. Asterisks represent non-TCGA cohorts. (b) Points show mean sensitivity for detecting mutations with deTiN (red) and without deTiN (blue). Means were derived from 256 of the 304 tumors that were matched with both a tumor-adjacent and a blood normal sample and had a sufficient number of somatic events to robustly estimate TiN (TiN = 0 [n=230]; TiN=0.01 [n=9]; TiN = 0.03 [n=9]; TiN=0.07 [n=4]; TiN=0.15 [n=1]; TiN=0.17 [n=1]; TiN=0.74 [n=1]; TiN=0.94 [n=1]). Error bars indicate standard error. (c) Histology images of selected adjacent tissue samples with evidence supporting TiN (n=1 patient sample for each image and plot). deTiN aSCNA data supporting TiN estimate is displayed for top two samples; points indicate allele-fraction of heterozygous germline SNPs, blue (tumor) and red (normal) points are used for TiN estimation, and grey points are not used by deTiN. The bottom plot displays deTiN somatic variant data supporting the TiN estimate for the bottom sample. Points indicate allele-fraction of variants in the tumor (x-axis) and normal (y-axis) samples; error bars indicate 95% beta confidence intervals. The green asterisk represents the KRAS G12V mutation, red points represent SSNVs recovered by deTiN, blue points are called before deTiN, and grey points are rejected by deTiN and MuTect as germline or artifact. Each plot displays data supporting TiN from a single tumor-normal pair corresponding to the image on the left (n = 1). (d) Illustration of three modes of contamination. Posterior distribution functions for TiN based on aSCNA data are shown clustered (red and orange) and unclustered for individual events (dashed grey). In the mixture scenario, TiN has two possible values: the lower represents events unique to the tumor cells (red) and the higher represents events shared between the tumor cells and the sibling precursor cells (orange).
References
References for main text
-
- Saunders CT et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–7 (2012). - PubMed
Supplementary References:
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
