

Patterns of genomic site inheritance in HIV-1M inter-subtype recombinants delineate the most likely genomic sites of subtype-specific adaptation
- PMID: 29942655
- PMCID: PMC6007327
- DOI: 10.1093/ve/vey015
Patterns of genomic site inheritance in HIV-1M inter-subtype recombinants delineate the most likely genomic sites of subtype-specific adaptation
Abstract
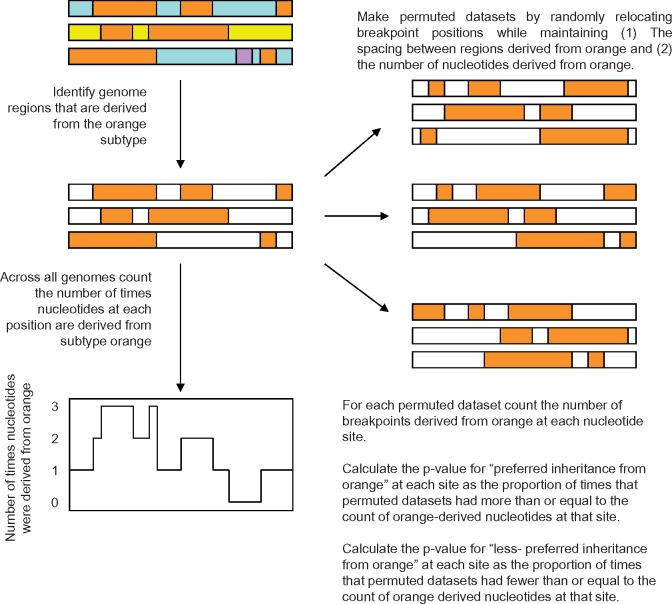
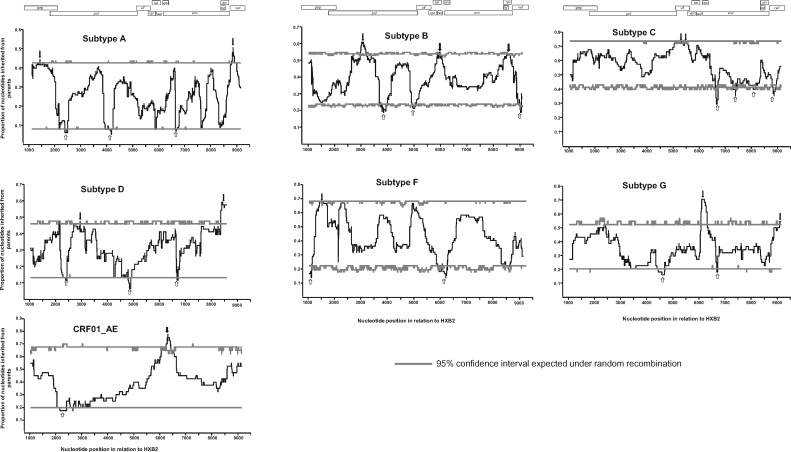
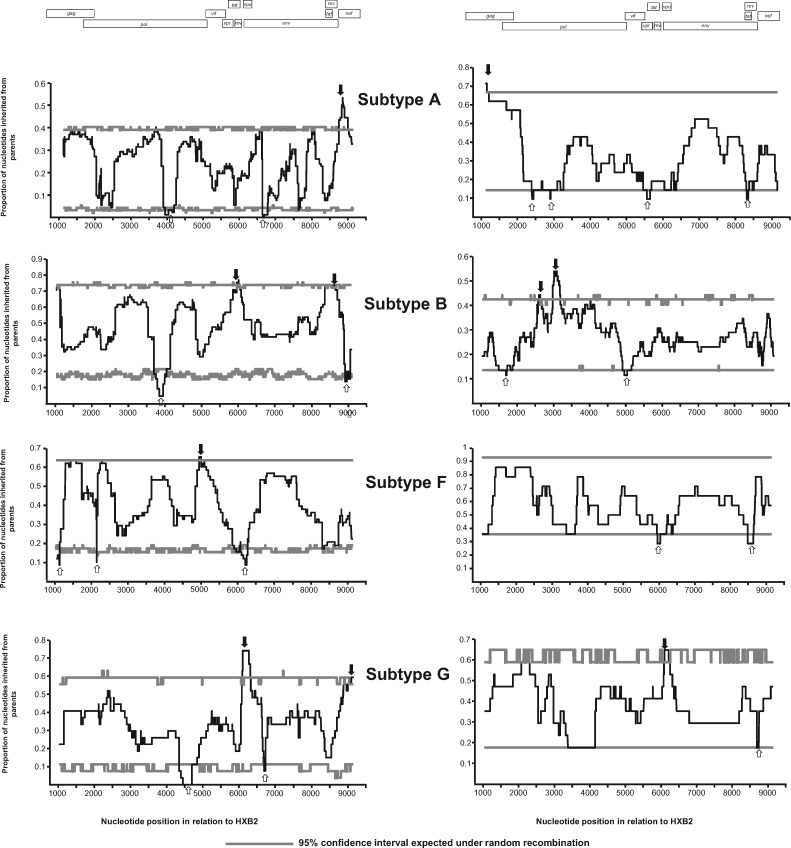
Recombination between different HIV-1 group M (HIV-1M) subtypes is a major contributor to the ongoing genetic diversification of HIV-1M. However, it remains unclear whether the different genome regions of recombinants are randomly inherited from the different subtypes. To elucidate this, we analysed the distribution within 82 circulating and 201 unique recombinant forms (CRFs/URFs), of genome fragments derived from HIV-1M Subtypes A, B, C, D, F, and G and CRF01_AE. We found that viruses belonging to the analysed HIV-1M subtypes and CRF01_AE contributed certain genome fragments more frequently during recombination than other fragments. Furthermore, we identified statistically significant hot-spots of Subtype A sequence inheritance in genomic regions encoding portions of Gag and Nef, Subtype B in Pol, Tat and Env, Subtype C in Vif, Subtype D in Pol and Env, Subtype F in Gag, Subtype G in Vpu-Env and Nef, and CRF01_AE inheritance in Vpu and Env. The apparent non-randomness in the frequencies with which different subtypes have contributed specific genome regions to known HIV-1M recombinants is consistent with selection strongly impacting the survival of inter-subtype recombinants. We propose that hotspots of genomic region inheritance are likely to demarcate the locations of subtype-specific adaptive genetic variations.
Keywords: CRF/URF; HIV-1; permutation-based test; recombination; selection.
Figures
Similar articles
-
Extensive and complex HIV-1 recombination between B', C and CRF01_AE among IDUs in south-east Asia.AIDS. 2012 Jun 1;26(9):1121-9. doi: 10.1097/QAD.0b013e3283522c97. AIDS. 2012. PMID: 22333750
-
HIV-1 diversity in infected individuals in Suzhou and Suqian, China.Springerplus. 2016 Jun 24;5(1):886. doi: 10.1186/s40064-016-2378-z. eCollection 2016. Springerplus. 2016. PMID: 27386334 Free PMC article.
-
The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin.J Virol. 1996 Oct;70(10):7013-29. doi: 10.1128/JVI.70.10.7013-7029.1996. J Virol. 1996. PMID: 8794346 Free PMC article.
-
Identification of two novel HIV-1 circulating recombinant forms of CRF111_01C and CRF116_0108 in southwestern Yunnan, China.Virulence. 2022 Dec;13(1):19-29. doi: 10.1080/21505594.2021.2010399. Virulence. 2022. PMID: 34951549 Free PMC article.
-
Current Trends of HIV Recombination Worldwide.Infect Dis Rep. 2013 Jun 6;5(Suppl 1):e4. doi: 10.4081/idr.2013.s1.e4. eCollection 2013 Jun 6. Infect Dis Rep. 2013. PMID: 24470968 Free PMC article. Review.
Cited by
-
Near full genome characterization of HIV-1 unique recombinant forms in Cameroon reveals dominant CRF02_AG and F2 recombination patterns.J Int AIDS Soc. 2019 Jul;22(7):e25362. doi: 10.1002/jia2.25362. J Int AIDS Soc. 2019. PMID: 31353798 Free PMC article.
-
Estimates of HIV-1 within-host recombination rates across the whole genome.Virus Evol. 2025 Jul 25;11(1):veaf052. doi: 10.1093/ve/veaf052. eCollection 2025. Virus Evol. 2025. PMID: 40740855 Free PMC article.
-
Elucidation of Early Evolution of HIV-1 Group M in the Congo Basin Using Computational Methods.Genes (Basel). 2021 Apr 2;12(4):517. doi: 10.3390/genes12040517. Genes (Basel). 2021. PMID: 33918115 Free PMC article. Review.
References
-
- Bagust T. J. et al. (2000) ‘Avian Infectious Laryngotracheitis’, Revue Scientifique et Technique (International Office of Epizootics), 19: 483–92. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
