

ncdDetect2: improved models of the site-specific mutation rate in cancer and driver detection with robust significance evaluation
- PMID: 29945188
- PMCID: PMC6330011
- DOI: 10.1093/bioinformatics/bty511
ncdDetect2: improved models of the site-specific mutation rate in cancer and driver detection with robust significance evaluation
Abstract
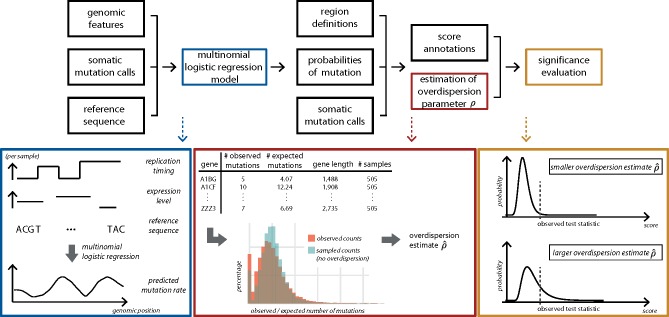
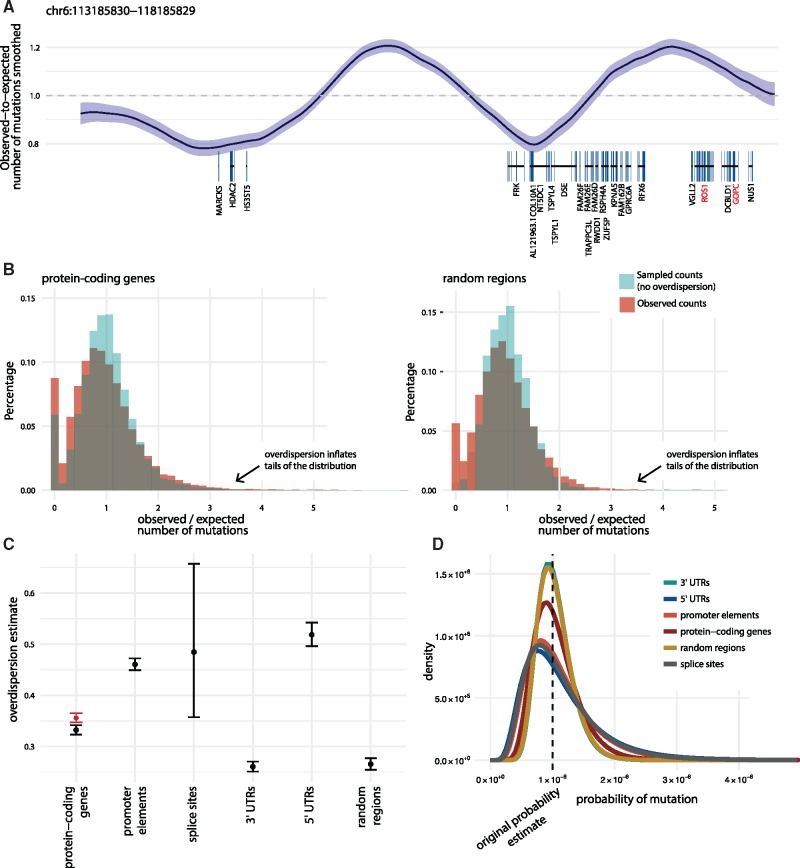
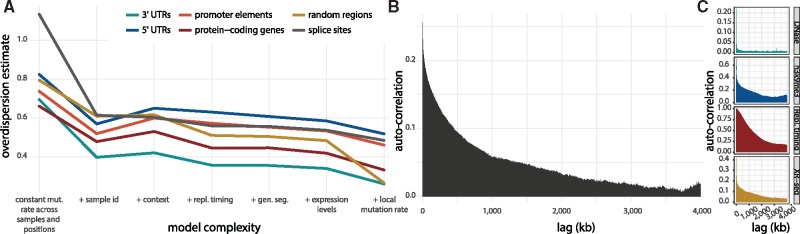
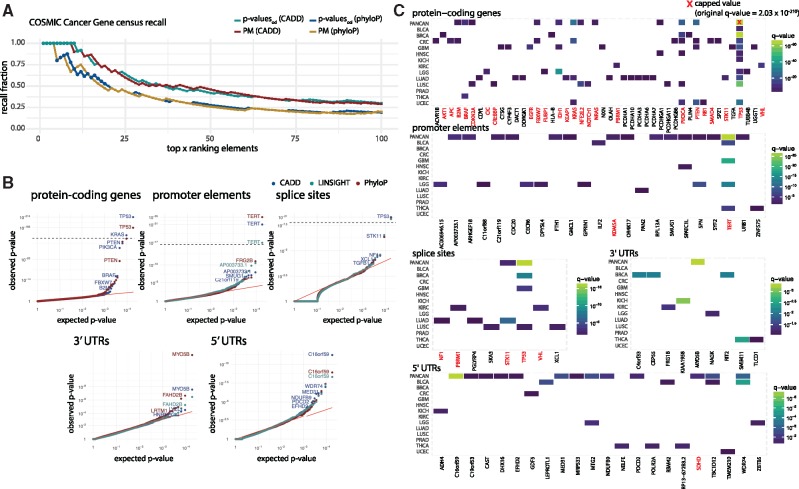
Motivation: Understanding the mutational processes that act during cancer development is a key topic of cancer biology. Nevertheless, much remains to be learned, as a complex interplay of processes with dependencies on a range of genomic features creates highly heterogeneous cancer genomes. Accurate driver detection relies on unbiased models of the mutation rate that also capture rate variation from uncharacterized sources.
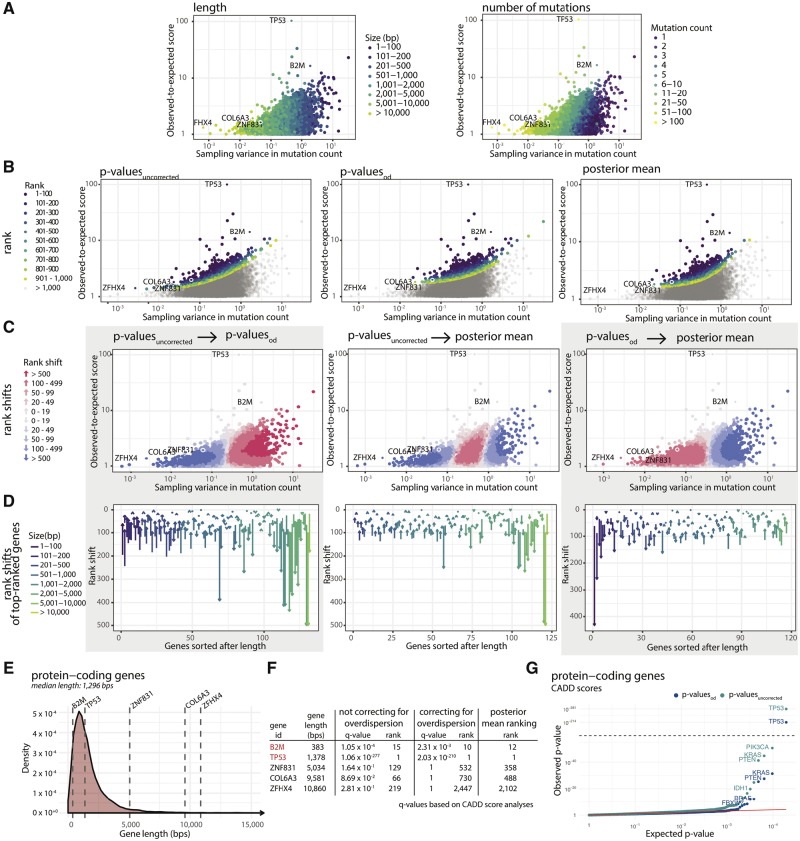
Results: Here, we analyse patterns of observed-to-expected mutation counts across 505 whole cancer genomes, and find that genomic features missing from our mutation-rate model likely operate on a megabase length scale. We extend our site-specific model of the mutation rate to include the additional variance from these sources, which leads to robust significance evaluation of candidate cancer drivers. We thus present ncdDetect v.2, with greatly improved cancer driver detection specificity. Finally, we show that ranking candidates by their posterior mean value of their effect sizes offers an equivalent and more computationally efficient alternative to ranking by their P-values.
Availability and implementation: ncdDetect v.2 is implemented as an R-package and is freely available at http://github.com/TobiasMadsen/ncdDetect2.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
Similar articles
-
A site specific model and analysis of the neutral somatic mutation rate in whole-genome cancer data.BMC Bioinformatics. 2018 Apr 19;19(1):147. doi: 10.1186/s12859-018-2141-2. BMC Bioinformatics. 2018. PMID: 29673314 Free PMC article.
-
Non-coding cancer driver candidates identified with a sample- and position-specific model of the somatic mutation rate.Elife. 2017 Mar 31;6:e21778. doi: 10.7554/eLife.21778. Elife. 2017. PMID: 28362259 Free PMC article.
-
Combined burden and functional impact tests for cancer driver discovery using DriverPower.Nat Commun. 2020 Feb 5;11(1):734. doi: 10.1038/s41467-019-13929-1. Nat Commun. 2020. PMID: 32024818 Free PMC article.
-
MEScan: a powerful statistical framework for genome-scale mutual exclusivity analysis of cancer mutations.Bioinformatics. 2021 Jun 9;37(9):1189-1197. doi: 10.1093/bioinformatics/btaa957. Bioinformatics. 2021. PMID: 33165532 Free PMC article.
-
Cancer driver gene discovery through an integrative genomics approach in a non-parametric Bayesian framework.Bioinformatics. 2017 Feb 15;33(4):483-490. doi: 10.1093/bioinformatics/btw662. Bioinformatics. 2017. PMID: 27797769 Free PMC article.
Cited by
-
Gsw-fi: a GLM model incorporating shrinkage and double-weighted strategies for identifying cancer driver genes with functional impact.BMC Bioinformatics. 2024 Mar 6;25(1):99. doi: 10.1186/s12859-024-05707-8. BMC Bioinformatics. 2024. PMID: 38448819 Free PMC article.
-
Non-coding driver mutations in human cancer.Nat Rev Cancer. 2021 Aug;21(8):500-509. doi: 10.1038/s41568-021-00371-z. Epub 2021 Jul 6. Nat Rev Cancer. 2021. PMID: 34230647 Review.
-
DeepAlloDriver: a deep learning-based strategy to predict cancer driver mutations.Nucleic Acids Res. 2023 Jul 5;51(W1):W129-W133. doi: 10.1093/nar/gkad295. Nucleic Acids Res. 2023. PMID: 37078611 Free PMC article.
-
Identifying somatic driver mutations in cancer with a language model of the human genome.Comput Struct Biotechnol J. 2025 Jan 17;27:531-540. doi: 10.1016/j.csbj.2025.01.011. eCollection 2025. Comput Struct Biotechnol J. 2025. PMID: 39968174 Free PMC article.
-
MutSpot: detection of non-coding mutation hotspots in cancer genomes.NPJ Genom Med. 2020 Jun 5;5:26. doi: 10.1038/s41525-020-0133-4. eCollection 2020. NPJ Genom Med. 2020. PMID: 32550006 Free PMC article.
References
-
- Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodological), 57, 289–300.
-
- Box G.E. et al. (2015) Time Series Analysis: Forecasting and Control. John Wiley & Sons Inc., Hoboken, New Jersey, USA.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
