

Trisomy of human chromosome 21 enhances amyloid-β deposition independently of an extra copy of APP
- PMID: 29945247
- PMCID: PMC6061702
- DOI: 10.1093/brain/awy159
Trisomy of human chromosome 21 enhances amyloid-β deposition independently of an extra copy of APP
Erratum in
-
Corrigendum.Brain. 2019 Jun 1;142(6):e25. doi: 10.1093/brain/awz061. Brain. 2019. PMID: 30879033 Free PMC article. No abstract available.
Abstract
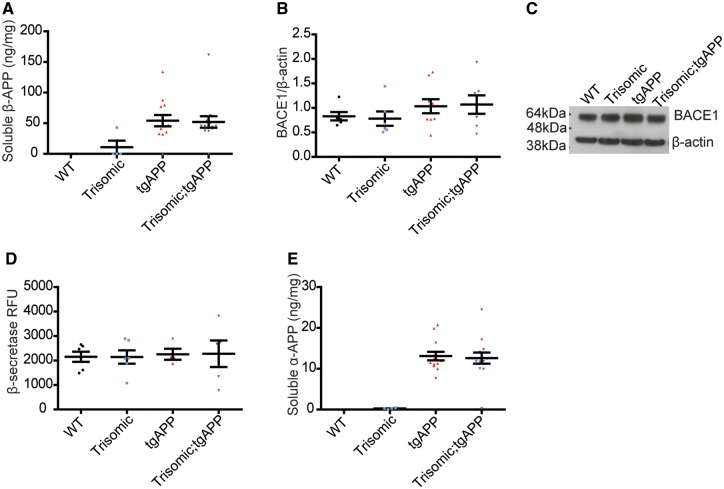
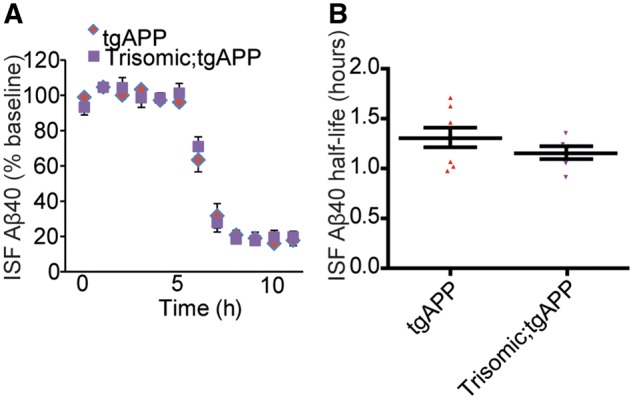
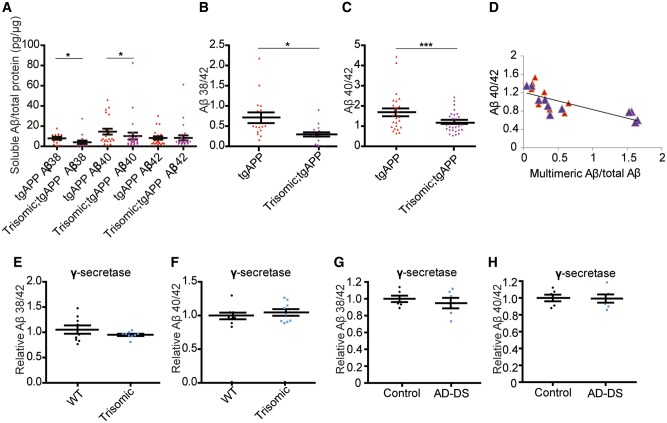
Down syndrome, caused by trisomy of chromosome 21, is the single most common risk factor for early-onset Alzheimer's disease. Worldwide approximately 6 million people have Down syndrome, and all these individuals will develop the hallmark amyloid plaques and neurofibrillary tangles of Alzheimer's disease by the age of 40 and the vast majority will go on to develop dementia. Triplication of APP, a gene on chromosome 21, is sufficient to cause early-onset Alzheimer's disease in the absence of Down syndrome. However, whether triplication of other chromosome 21 genes influences disease pathogenesis in the context of Down syndrome is unclear. Here we show, in a mouse model, that triplication of chromosome 21 genes other than APP increases amyloid-β aggregation, deposition of amyloid-β plaques and worsens associated cognitive deficits. This indicates that triplication of chromosome 21 genes other than APP is likely to have an important role to play in Alzheimer's disease pathogenesis in individuals who have Down syndrome. We go on to show that the effect of trisomy of chromosome 21 on amyloid-β aggregation correlates with an unexpected shift in soluble amyloid-β 40/42 ratio. This alteration in amyloid-β isoform ratio occurs independently of a change in the carboxypeptidase activity of the γ-secretase complex, which cleaves the peptide from APP, or the rate of extracellular clearance of amyloid-β. These new mechanistic insights into the role of triplication of genes on chromosome 21, other than APP, in the development of Alzheimer's disease in individuals who have Down syndrome may have implications for the treatment of this common cause of neurodegeneration.
Figures
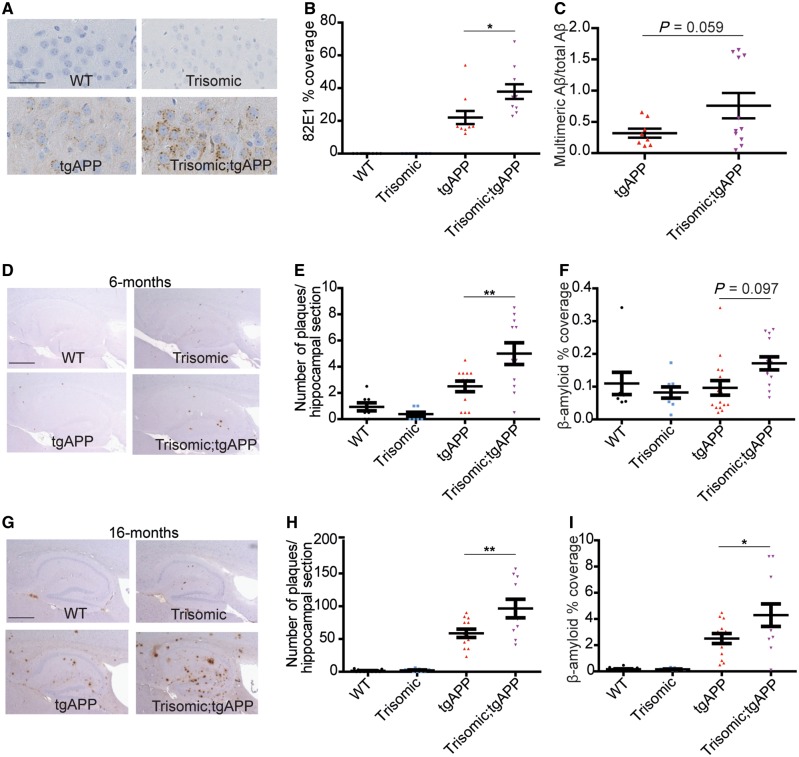
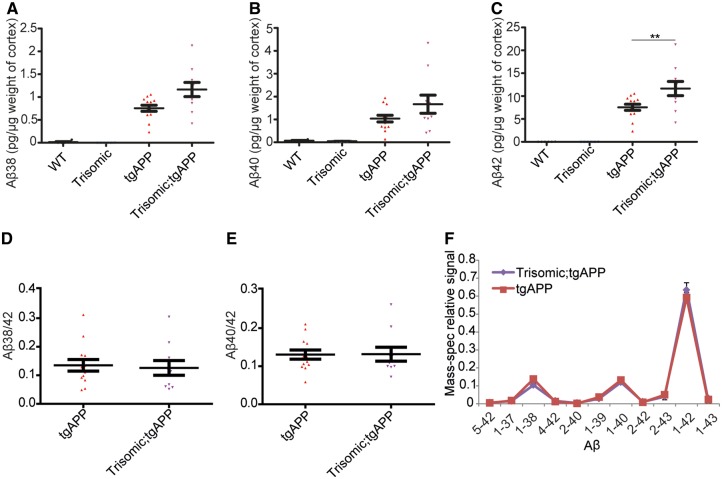
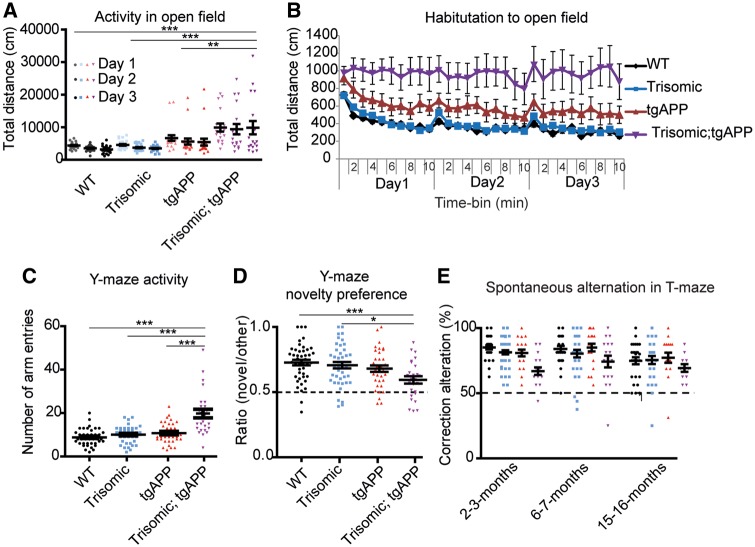
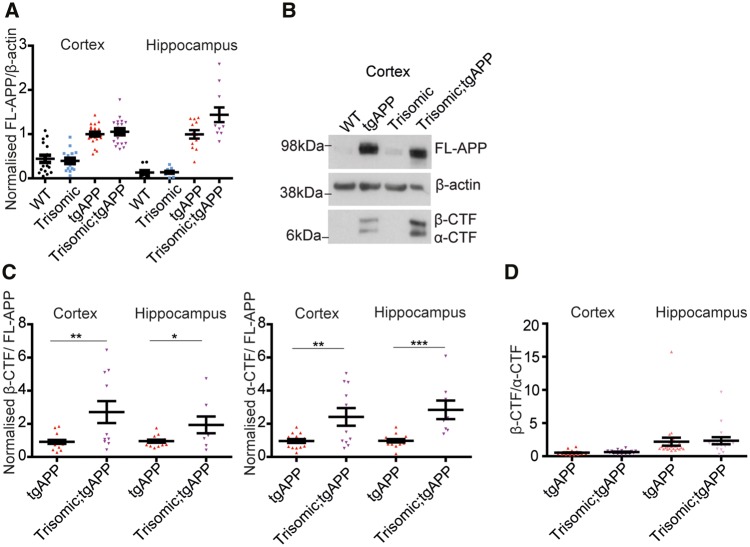
Comment in
-
New clues to the genetic links between AD and Down syndrome.Nat Rev Neurol. 2018 Sep;14(9):508-509. doi: 10.1038/s41582-018-0045-4. Nat Rev Neurol. 2018. PMID: 29991821 No abstract available.
References
-
- Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M et al. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging 2004; 25: 1263–72. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
